همترازسازی ساختاری
همترازسازی ساختاری، تلاشی جهت یافتن تشابه در ساختار فضایی دو یا چند پلیمر است. این فرایند معمولاً روی ساختار سوم پروتئین اعمال میشود اما میتواند روی مولکولهای بزرگ RNA نیز انجام شود. همترازسازی ساختاری ابزاری مناسب برای مقایسه میان پروتئینهایی است که دنباله آمینواسیدی آنها، شباهت کمی با یکدیگر دارد. جایی که تکنیکهای مبتنی بر بررسی دنبالهها برای تطابق پروتئینها نمیتواند شباهتها و روابط تکاملی میان ساختارها را به سادگی پیدا کند.
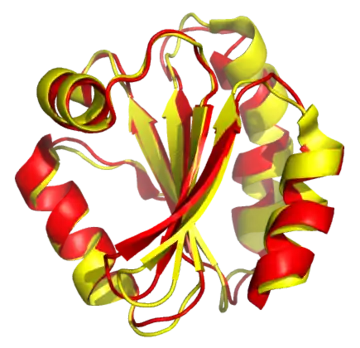
هم ترازسازی ساختاری میتواند برای مقایسه دو یا بیشتر از دو ساختار استفاده شود. از آنجایی که این روش از اطلاعات سه بعدی ساختارها استفاده میکند، این روش تنها روی ساختارهایی قابل اجرا است که شکل سه بعدی آنها بهطور کامل شناخته شده باشد. ساختارهای فضایی معمولاً به وسیله بلورشناسی پرتو ایکس یا طیفسنجی تشدید مغناطیسی هستهای به دست میآیند.
به کمک این روش، این امکان وجود دارد که میان ساختار دقیق فضایی یک پروتئین و ساختار پیشبینی شده آن توسط روشهای پیشبینی ساختار پروتئین، همترازسازی ساختاری انجام داد. از نتیجه این آزمایش میتوان میزان دقت و درستی ساختارهای پیشبینی شده را ارزیابی کرد.[1]
خروجی یک تراز ساختاری، یک ترکیب از مجموعههای مختصات اتمی و حداقل انحراف مقدار مؤثر بین ساختار هاست. حداقل انحراف مقدار مؤثر میزان واگرایی دو ساختار نسبت به یکدیگر را معین میکند، به این معنی که هر چه این اختلاف کمتر باشد، دو ساختار به یکدیگر شبیهترند و در غیر اینصورت تفاوت بیشتری با یکدیگر دارند.
دادههای تولید شده به وسیله همترازسازی ساختاری
کمترین داده تولید شده پس از این فرایند، مجموعه اجزائی است که میان ساختارها مشابه شناخته شدهاست. این مجموعه برای یافتن شکل سه بعدی این ساختارها در کنار یکدیگر، در حالت تراز شده استفاده میشود. پس از یافتن شکل سه بعدی تراز شده ساختارها، میتوان میزان انحراف مقدار مؤثر میان ساختارها را محاسبه کرد. همچنین میتوان محاسبات پیچیدهتری از قبیل تست فاصله کلی را انجام داد.[2]
همترازسازی ساختاری، میتواند به عنوان همترازساز دنبالهای نیز استفاده شود، چرا که نقاطی که شکل سه بعدی ساختارها تطابق داشتهاند، یعنی در دنباله تشکیل دهندهشان نیز تشابه وجود داشتهاست. ضمناً به کمک درصد اجزایی که در شکل فضایی با یکدیگر تطابق دارند میتوان معیاری جهت مقایسه اینکه دو ساختار چقدر شبیه هستند، انجام داد.
پیچیدگی الگوریتمی
الگوریتم بهینه
اثبات شدهاست که یافتن یک تراز ساختاری بهینه برای چند پروتئین مسئلهای NP کامل است.[3] البته این موضوع به این معنا نیست که این مسئله حل ناشدنی است، بلکه به کمک مقیاسهای اندازهگیری که تعریف شدهاند الگوریتمهایی موجودند که میتوانند ترازی با دقت قابل قبول به دست بیاورند.[4] زمان اجرای الگوریتمها نه تنها وابسته به تعداد واحدها بلکه به شکل ذاتی پروتئینها نیز وابسته است.
الگوریتمهای تقریبی
الگوریتمهایی چندجملهای زیادی توسعه یافتهاند که میتوانند جوابهایی تقریباً بهینه پیدا کنند. البته این الگوریتمها نیز جهت بررسی پروتئینها و ساختارهای پیچیده بسیار زمانبر و هزینهبر هستند؛ بنابراین در واقع الگوریتمهای عملیاتی زیادی که بتوانند به شکل گسترده استفاده بشوند، وجود ندارد. ضمناً اکثر این الگوریتمها ابتکاری هستند. در سالهای اخیر الگوریتمهایی جهت یافتن ساختارهای بهینه محلی، که میتوانند به شکلی سریع و عملیاتی پاسخ بدهند، توسعه یافتهاند.[5]
شیوهها
از شیوههای مختلف همترازسازی ساختاری جهت تولید پایگاههای داده همگانی استفاده شدهاست. به طوری برای هر دو پروتئین موجود، تراز ساختاری میان آنها وجود داشته باشد. در طی سالیان اخیر این پایگاههای داده بهتر و کاملتر شدهاند.
DALI
یک شیوه محبوب برای همترازسازی استفاده از روش همترازسازی به کمک ماتریس فاصله است. این روش هر پروتئین ورودی را به اجزایی کوچک میشکند و فاصله میان این اجزا را برای هر ساختار در یک ماتریس ذخیره میکند. نهایتاً سعی میکند برای تراز پروتئینها، طوری اجزای کوچکشان را با یکدیگر تطابق دهد که ماتریس فاصلهها تا حد امکان درست و صحیح برقرار بمانند. یعنی اجزایی را با یکدیگر تطابق میدهد که فاصلهشان در تمامی ساختارها تقریباً برابر باشد.[6]
از این شیوه در تولید پایگاه داده FSSP استفاده شدهاست که برای مقایسه ساختار پروتئینها استفاده میشود.
CE
این شیوه نیز مشابه روش قبل، پروتئین را به اجزای کوچکتر تقسیم میکند و سعی میکنند یک جفتکردن بهینه بین اجزا پیدا کند. این روش سعی میکند خصوصیات محیطی محلی، ساختار دوم پروتئین و الگوهای پیوندهای هیدروژنی را نیز در نظر بگیرد و بر اساس آنها تراز را انجام دهد.[7]
SSAP
این روش از برنامهنویسی پویا استفاده میکند تا یک تراز ساختاری میان پروتئینها پیدا کند و سعی میکند تا پروتئینها را اتم به اتم در یک فضای سهبعدی مطابقت بدهد. این روش برای یافتن اجزای کوچکترش، از شیوهای متفاوت با دو روش قبلی استفاده میکند. این شیوه در ابتدا صرفاً برای تراز کردن دو ساختار استفاده میشد اما امروزه برای همترازسازی چند پروتئین نیز استفاده میشود.[8]
به کمک این روش پایگاه داده CATH توسعه یافتهاست.
همترازسازی ساختار در RNA
تکنیکهای همترازسازی ساختاری به صورت سنتی در میان پروتئینها رایج بودهاست. با این وجود رشتههای RNA نیز به جهت پیوندهای هیدروژنی که میان نوکلئوتیدهایش شکل میگیرد؛ ساختاری سه بعدی در فضا تشکیل میدهند؛ بنابراین بررسی ساختار RNAهایی که در سلولها ترجمه نمیشوند و خودشان درون سلول کاربرد دارند، میتواند حائز اهمیت باشد. در سالهای اخیر روشهایی برای تراز ساختاری در میان مولکولهای RNA توسعه یافتهاست.[9]
جستارهای وابسته
منابع
- Zhang Y, Skolnick J (2005). "The protein structure prediction problem could be solved using the current PDB library". Proc Natl Acad Sci USA. 102 (4): 1029–34. doi:10.1073/pnas.0407152101. PMC 545829. PMID 15653774.
- Zemla A. (2003). "LGA — A Method for Finding 3-D Similarities in Protein Structures". Nucleic Acids Research. 31 (13): 3370–3374. doi:10.1093/nar/gkg571. PMC 168977. PMID 12824330.
- Lathrop RH. (1994). "The protein threading problem with sequence amino acid interaction preferences is NP-complete". Protein Eng. 7 (9): 1059–68. CiteSeerX 10.1.1.367.9081. doi:10.1093/protein/7.9.1059. PMID 7831276.
- Poleksic A (2009). "Algorithms for optimal protein structure alignment". Bioinformatics. 25 (21): 2751–2756. doi:10.1093/bioinformatics/btp530. PMID 19734152.
- Martinez L, Andreani, R, Martinez, JM. (2007). "Convergent algorithms for protein structural alignment". BMC Bioinformatics. 8: 306. doi:10.1186/1471-2105-8-306. PMC 1995224. PMID 17714583.
- Holm L, Sander C (1996). "Mapping the protein universe". Science. 273 (5275): 595–603. doi:10.1126/science.273.5275.595. PMID 8662544.
- Shindyalov, I.N.; Bourne P.E. (1998). "Protein structure alignment by incremental combinatorial extension (CE) of the optimal path". Protein Engineering. 11 (9): 739–747. doi:10.1093/protein/11.9.739. PMID 9796821.
- Taylor WR, Flores TP, Orengo CA (1994). "Multiple protein structure alignment". Protein Sci. 3 (10): 1858–70. doi:10.1002/pro.5560031025. PMC 2142613. PMID 7849601.
- Torarinsson E, Sawera M, Havgaard JH, Fredholm M, Gorodkin J (2006). "Thousands of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA structure". Genome Res. 16 (7): 885–9. doi:10.1101/gr.5226606. PMC 1484455. PMID 16751343.