بازسازی توالی
بازسازی توالی (به انگلیسی Sequence Assembly) در بیوانفورماتیک، به ادغام و همتراز کردن قسمتهای مختلف توالی DNA برای بازسازی توالی اصلی اطلاق میشود. اهمیت بازسازی توالی در آن است که فناوریهای موجود برای تعیین توالی DNA قادر به خواندن تمام ژنوم در یک مرحله نیستند و توالی به صورت قطعههای ۲۰ تا ۳۰۰۰۰ حرفی (هر حرف نشانهٔ یک باز است) خوانده میشود، این قطعهها معمولاً نتیجهٔ رونویسی ژن (ESTها) یا تعیین توالی ژنوم با روش شاتگاناند.[1]
پیچیدگیهای این مسئله
میتوان مسئلهٔ بازسازی توالی DNA را به این تشبیه کرد که؛ از یک کتاب نسخههای مختلفی ایجاد کنیم، هر نسخه را با دستگاهی متفاوت قطعه قطعه کنیم، تغییراتی در قطعههای ایجاد شده بدهیم، قطعاتی از کتابهای دیگر به آنها اضافه کنیم و برخی قطعهها را نابود کنیم و در نهایت بخواهیم با دیدن قطعههای نهایی، کتاب اصلی را بازسازی کنیم. مسئله حتی پیچیدهتر میشود وقتی این کتاب شامل نوشتهها و قطعات تکراری بوده باشد.[1]
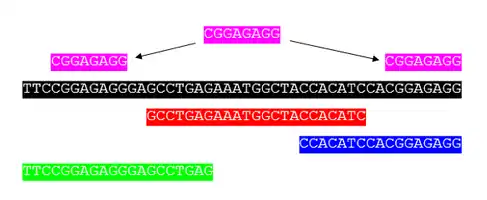
تاریخچهٔ بازسازی ژنوم
اولین روشها
در اواخر دههٔ ۱۹۸۰ و اوایل دههٔ ۱۹۹۰ میلادی، اولین برنامههای بازسازی توالی به عنوان نسخههای پیشرفتهتر از برنامههای سادهٔ همترازسازی معرفی شدند تا توالیهایی که دستگاههای تعیین توالی قطعاتی از آنها را یافته بودند، بازسازی کنند.[1]
با پیشرفت علم و فناوری، در ابتدا ژنوم ویروسهای ساده، سپس باکتریها و در نهایت یوکاریوتها مورد بررسی قرار گرفتند، و پیچیدگی مسئلهٔ بازسازی ژنوم با افزایش طول و پیچیدگی توالیهای یافته شده بیشتر شد، پیچیدگیهایی از قبیل[1];
- پردازش حجم عظیمی از دادهها (از مرتبهٔ ترابایت) که نیاز به رایانش خوشهای دارد،
- قسمتهای تکراری ژنوم که میتوانند پیچیدگی زمانی و مصرف حافظهٔ الگوریتمها را، در بدترین حالت، به صورت نمایی افزایش دهند،
- خطا در خواندن توالیها که میتواند موجب پیچیدگی و خطا در بازسازی شود.
دانشمندان برای بازسازی توالیهای پیچیدهٔ ژنوم یوکاریوتهایی چون مگس سرکه (در سال ۲۰۰۰ میلادی) و انسان (در سال ۲۰۰۱ میلادی) ابزارهای بازسازی توالیای همچون[2] Celera Assembler و[3] Arachne را ارائه کردند که قادر به بازسازی توالیهایی به طول ۱۰۰ تا ۳۰۰ میلیون حرفاند. پیروی این ابداعات، در مراکز بازسازی توالیهای ژنوم، گروههایی به ساخت ابرابزارهای بازسازی پرداختند که نمونهٔ متن بازی از آن[4] AMOS است که تلاشی برای گردآوری دستیافتههای مختلف در این زمینه بود.[1]
تأثیرات تغییر فناوری
پیچیدگی مسئلهٔ بازسازی توالی تحت تأثیر تعداد توالی و طول هرکدام است. تعداد زیادی توالی طولانی به یافتن بهتر همپوشانیها کمک میکند، ولی زمان اجرای الگوریتمها به صورت نمایی با تعداد و طول توالیها افزایش مییابد. از طرفی، توالیهای کوتاهتر به راحتی همتراز میشوند، اما موجب پیچیدگی بازسازی میشوند، چراکه یافتن و استفاده از قسمتهای تکراری در قطعات کوتاهتر، سختتر است.[1]
در ابتدا، تعداد کمی توالی کوتاه از ژنوم به سختی و پس از چند هفته تلاش در آزمایشگاه بدست میآمد که به راحتی و به صورت دستی (!) همتراز میشدند.
با ابداع روش سنگر در سال ۱۹۷۵ میلادی و تا سال ۲۰۰۰ میلادی، فناوری به جایی رسید که ابزارهای پیشرفته و خودکار به صورت شبانهروزی توالیهای ژنوم را بدست آورند و در نتیجهٔ آن نیاز به روشهایی برای پردازش توالیهایی ایجاد شده که[1];
- حدود ۸۰۰–۹۰۰ حرف (باز) طول دارند،
- شامل قسمتهای ابداعی مانند توالییابی یا ارگانیسمهای تولید مثلکننده (Cloning Vectors) اند،
- مرتبهٔ خطای ۰/۵ تا ۱۰٪ دارند.
فناوری سنگر این امکان را ایجاد کرد که با تنها یک رایانه بتوان توالیهای باکتری با ۲۰۰۰۰ تا ۲۰۰۰۰۰ قطعه را بازسازی کرد، اما بازسازی توالیهای بزرگتر مانند ژنوم انسان (با حدود ۳۵ میلیون قطعه) همچنان نیاز به استفاده از پردازش موازی با چندین رایانه دارند.
شرکت بیوانفورماتیک 454Life Sciences در سالهای ۲۰۰۴/۲۰۰۵ میلادی روش Pyroscequencing را معرفی کرد که قطعههای کوتاهتری نسبت به روش سنگر ایجاد میکرد (در ابتدا قطعاتی به طول حدود ۱۰۰ حرف و در حال حاضر ۴۰۰–۵۰۰ حرف)، و توان عملیاتی و سرعت بالای آن موجب شد مورد استفادهٔ مراکز بازسازی توالی قرار گیرد.[5]
حجم دادهها و خطاهای ابزارهای توالی یابی در خواندن قطعات موجب تأخیر در ساخت ابزارهای بازسازی توالی شد و در ابتدا (سال ۲۰۰۴ میلادی) فقط ابزار Newbler از شرکت ۴۵۴ در دسترس بود. اولین ابزار در دسترس که میتوانست علاوه بر قطعات ۴۵۴، ترکیب قطعات ۴۵۴ و سنگر را برای بازسازی ادغام کند، نسخهٔ ترکیبی ابزار MIRA بود که Chevreux و همکارانش در اواسط سال ۲۰۰۷ میلادی ارائه کردند و پس از آن به بازسازی توالی از توالیهای یافته شده با فناوری هایمختلف، بازسازی ترکیبی (Hybrid Assembly) اطلاق شد.[1]
فناوری ایلومینا از سال ۲۰۰۶ میلادی در دسترس است که میتواند در هر اجرا حدود ۱۰۰ میلیون قطعه را، در یک دستگاه، بخواند (ژنوم انسان نیاز به خواندن حدود ۳۵ میلیون قطعه دارد) که در ابتدا طول این قطعات به ۳۶ حرف (باز) محدود بود که برای روش De-Novo مناسب نبود، اما هماکنون طول این قطعات به بالای ۱۰۰ حرف میرسد.[1]
ابزار بازسازی[6] SHARCGS که اواخر سال ۲۰۰۷ میلادی معرفی شد، اولین ابزار بازسازی توالی بود که از قطعات توالی بدست آمده با Solexa (نام قبلی ایلومینا) استفاده میکرد که پس از آن چندین ابزار دیگر نیز ارائه شدند.[1]
از ابزارها و فناوریهای جدیدتر در این زمینه میتوان به SOLiD, Ion Torrent, SMRT و توالییابی Nanopore اشاره کرد.
تفاوت با بازسازی EST
بازسازی برچسب توالی بیان شده (Expressed Sequence Tag) از جهات مختلفی با بازسازی توالی ژنوم متفاوت است[1];
- توالیهایی که در EST بازسازی میشوند، قطعات مختلف mRNA هستند که بیانگر بخشی از ژنوم (قسمتهایی از اگزونها) اند ولی در بازسازی ژنوم تمام توالی DNA بدست میآید.
- توالیهای mRNA نسبت به ژنوم قسمتهای تکراری کمتری دارند (قسمتهای تکراری معمولاً در اینترونها دیده میشوند).
- برخی ژنها بیشتر از بقیه بیان میشوند (ژنهای خانهبان) که موجب وجود توالیهای تکراری در دادهها میشود.
- گاهی ژنها همپوشانی دارند اما باید جداگانه بازسازی شوند.

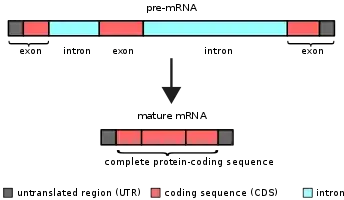
بازسازی EST خود پیچیدگیهایی نظیر پیرایشهای دگرسان (alternative splicing)، تراپیرایش، چندریختی تک-نوکلئوتید و تغییرات پس از رونویسی (Post-transcription modification) دارد.[1]
انواع بازسازی توالی
بازسازی توالی ژنوم به دو صورت مختلف انجام میگیرد:
۱. De-Novo: بازسازی تمام توالی از قسمتهای کوچکتر آن.[7]
این روش بسیار کندتر است و حافظهٔ بیشتری مصرف میکند، چراکه تمامی قطعات باید با هم مقایسه شوند (که در حالت عادی از مرتبهٔ زمانی است و البته با استفاده از جدول درهمسازی بهبود مییابد).

۲. استفاده از یک توالی اولیه و تطبیق قطعههای بدست آمده با قسمتهای مختلف آن (mapping) برای ایجاد توالیای جدید که مشابه توالی اولیه، ولی نه الزاماً یکسان با آن، است.[1]
اگر از نوع توالی پیش زمینهای داشته باشیم، لازم نیست از ابتدا تمام توالی را بازسازی کنیم و با داشتن یک توالی به عنوان قالب میتوانیم تفاوتهای جزئی آن با توالی مورد نظر را با استفاده از قطعات بدست آمده اصلاح کنیم.
الگوریتم حریصانه
این مسئله مشابه یافتن کوتاهترین ابردنبالهٔ مشترک (Shortest Common Supersequence) برای رشتههای داده شدهاست که راه حل حریصانهی آن بدین صورت است:[1]
- همترازی دوبهدوی تمامی قطعات (رشتههای داده شده) را بیآب.
- دو قطعه که بیشترین همپوشانی را دارند انتخاب کن.
- قطعات انتخاب شده را ادغام کن.
- مراحل ۲ و ۳ را تکرار کن تا تنها یک قطعه باقی بماند.
- قطعهٔ باقیمانده را به عنوان جواب مسئله گزارش کن.
البته این جواب الزاماً بهینه نیست.
ابزارهای در دسترس
ابزارهای بازسازی توالی که توانایی بازسازی De-Novo را در حداقل یکی از فناوریهای پشتیبانی شده دارند در جدول زیر معرفی شدهاند:[1]
| نام | نوع استفاده | فناوریها | گردآورنده | تاریخ ارائه /
تاریخ آخرین تغییر |
مجوز* | صفحهٔ خانگی |
|---|---|---|---|---|---|---|
| ABySS | large (genomes) | Solexa, SOLiD | Simpson, J. et al. | ۲۰۰۸ / ۲۰۱۴ | NC-A | link |
| ALLPATHS-LG | (large) genomes | Solexa, SOLiD | Gnerre, S. et al. | ۲۰۱۱ | OS | link |
| AMOS | genomes | Sanger, 454 | Salzberg, S. et al. | ۲۰۰۲? / ۲۰۱۱ | OS | link |
| Arapan-M | Medium Genomes (e.g. E.coli) | All | Sahli, M. & Shibuya, T. | ۲۰۱۱ / ۲۰۱۲ | OS | link |
| Arapan-S | Small Genomes (Viruses and Bacteria) | All | Sahli, M. & Shibuya, T. | ۲۰۱۱ / ۲۰۱۲ | OS | link |
| Celera WGA Assembler / CABOG | (large) genomes | Sanger, 454, Solexa | Myers, G. et al. ; Miller G. et al. | ۲۰۰۴ / ۲۰۱۵ | OS | link |
| CLC Genomics Workbench & CLC Assembly Cell | genomes | Sanger, 454, Solexa, SOLiD | CLC bio | ۲۰۰۸ / ۲۰۱۰ / ۲۰۱۴ | C | link |
| Cortex | genomes | Solexa, SOLiD | Iqbal, Z. et al. | ۲۰۱۱ | OS | link |
| DBG2OLC | (large) genomes | Illumina, PacBio, Oxford Nanopore | Ye, C. et al | ۲۰۱۴/۲۰۱۶ | OS | link |
| DNA Baser Assembler | (small) genomes | Sanger, 454 | Heracle BioSoft SRL | ۰۴٫۲۰۱۶ | C | www.DnaBaser.com |
| DNA Dragon | genomes | Illumina, SOLiD, Complete Genomics, 454, Sanger | SequentiX | ۲۰۱۱ | C | link |
| DNAnexus | genomes | Illumina, SOLiD, Complete Genomics | DNAnexus | ۲۰۱۱ | C | link |
| DNASTAR Lasergene Genomics Suite | (large) genomes, exomes, transcriptomes, metagenomes, ESTs | Illumina, ABI SOLiD, Roche 454, Ion Torrent, Solexa, Sanger | DNASTAR | ۲۰۰۷ / ۲۰۱۶ | C | link |
| Edena | genomes | Illumina | D. Hernandez, P. François, L. Farinelli, M. Osteras, and J. Schrenzel. | ۲۰۰۸/۲۰۱۳ | OS | link |
| Euler | genomes | Sanger, 454 (,Solexa ?) | Pevzner, P. et al. | ۲۰۰۱ / ۲۰۰۶? | (C / NC-A?) | link |
| Euler-sr | genomes | 454, Solexa | Chaisson, MJ. et al. | ۲۰۰۸ | NC-A | link |
| Fermi | (large) genomes | Illumina | Li, H. | ۲۰۱۲ | OS | link |
| Forge | (large) genomes, EST, metagenomes | 454, Solexa, SOLID, Sanger | Platt, DM, Evers, D. | ۲۰۱۰ | OS | link |
| Geneious | genomes | Sanger, 454, Solexa, Ion Torrent, Complete Genomics, PacBio, Oxford Nanopore, Illumina | Biomatters Ltd | ۲۰۰۹ / ۲۰۱۳ | C | link |
| Graph Constructor | (large) genomes | Sanger, 454, Solexa, SOLiD | Convey Computer Corporation | ۲۰۱۱ | C | link |
| HINGE | genomes | PacBio/Oxford Nanopore | Kamath, Shomorony, Xia et. al.[8] | ۲۰۱۶ | OS | Software, Paper, Analyses |
| IDBA (Iterative De Bruijn graph short read Assembler) | (large) genomes | Sanger,454,Solexa | Yu Peng, Henry C. M. Leung, Siu-Ming Yiu, Francis Y. L. Chin | ۲۰۱۰ | (C / NC-A?) | link |
| LIGR Assembler (derived from TIGR Assembler) | genomic | Sanger | - | ۲۰۰۹/ ۲۰۱۲ | OS | link |
| MaSuRCA (Maryland Super Read - Celera Assembler) | (large) genomes | Sanger, Illumina, 454 | Aleksey Zimin, Guillaume Marçais, Daniela Puiu, Michael Roberts, Steven L. Salzberg, James A. Yorke | ۲۰۱۲ / ۲۰۱۳ | OS | link |
| MIRA (Mimicking Intelligent Read Assembly) | genomes, ESTs | Sanger, 454, Solexa | Chevreux, B. | ۱۹۹۸ / ۲۰۱۴ | OS | link |
| NextGENe | (small genomes?) | 454, Solexa, SOLiD | Softgenetics | ۲۰۰۸ | C | link |
| Newbler | genomes, ESTs | 454, Sanger | 454/Roche | ۲۰۰۴/۲۰۱۲ | C | link |
| PADENA | genomes | 454, Sanger | 454/Roche | ۲۰۱۰ | OS | link |
| PASHA | (large) genomes | Illumina | Liu, Schmidt, Maskell | ۲۰۱۱ | OS | link |
| Phrap | genomes | Sanger, 454, Solexa | Green, P. | ۱۹۹۴ / ۲۰۰۸ | C / NC-A | link |
| TIGR Assembler | genomic | Sanger | - | ۱۹۹۵ / ۲۰۰۳ | OS | link |
| Trinity | Transcriptomes | short reads (paired, oriented, mixed) Illumina, 454, Solid,... | Grabher, MG et al.[9] | ۲۰۱۱/۲۰۱۶ | OS | https://github.com/trinityrnaseq/trinityrnaseq/wiki |
| Ray[10] | genomes | Illumina, mix of Illumina and 454, paired or not | Sébastien Boisvert, François Laviolette & Jacques Corbeil. | ۲۰۱۰ | OS [GNU General Public License] | link |
| Sequencher | genomes | traditional and next generation sequence data | Gene Codes Corporation | ۱۹۹۱ / ۲۰۰۹ / ۲۰۱۱ | C | link |
| SGA | (large) genomes | Illumina, Sanger (Roche 454?, Ion Torrent?) | Simpson, J.T. et al. | ۲۰۱۱ / ۲۰۱۲ | OS | link |
| SHARCGS | (small) genomes | Solexa | Dohm et al. | ۲۰۰۷ / ۲۰۰۷ | OS | link |
| SOPRA | genomes | Illumina, SOLiD, Sanger, 454 | Dayarian, A. et al. | ۲۰۱۰ / ۲۰۱۱ | OS | link |
| SparseAssembler | (large) genomes | Illumina, 454, Ion torrent | Ye, C. et al. | ۲۰۱۲ / ۲۰۱۲ | OS | link |
| SSAKE | (small) genomes | Solexa (SOLiD? Helicos?) | Warren, R. et al. | ۲۰۰۷ / ۲۰۱۴ | OS | link |
| SOAPdenovo | genomes | Solexa | Luo, R. et al. | ۲۰۰۹ / ۲۰۱۳ | OS | link |
| SPAdes | (small) genomes, single-cell | Illumina, Solexa, Sanger, 454, Ion Torrent, PacBio, Oxford Nanopore | Bankevich, A et al. | ۲۰۱۲ / ۲۰۱۵ | OS | link |
| Staden gap4 package | BACs (, small genomes?) | Sanger | Staden et al. | ۱۹۹۱ / ۲۰۰۸ | OS | link |
| Taipan | (small) genomes | Illumina | Schmidt, B. et al. | ۲۰۰۹ / ۲۰۰۹ | OS | link |
| VCAKE | (small) genomes | Solexa (SOLiD?, Helicos?) | Jeck, W. et al. | ۲۰۰۷ / ۲۰۰۹ | OS | link |
| Phusion assembler | (large) genomes | Sanger | Mullikin JC, et al. | ۲۰۰۳ / ۲۰۰۶ | OS | link |
| Quality Value Guided SRA (QSRA) | genomes | Sanger, Solexa | Bryant DW, et al. | ۲۰۰۹ / ۲۰۰۹ | OS | link |
| Velvet | (small) genomes | Sanger, 454, Solexa, SOLiD | Zerbino, D. et al. | ۲۰۰۷ / ۲۰۱۱ | OS | link |
| *مجوز: OS = متن باز؛ C = تجاری؛ C / NC-A = تجاری، اما رایگان برای استفادهٔ غیر تجاری و آکادمیک؛ براکت = غیر مشخص اما احتمالاً همان C / NC-A | ||||||
جستارهای وابسته
منابع
- «Sequence Assembly - Wikipedia».
- Myers, E. W. ; Sutton, GG; Delcher, AL; Dew, IM; Fasulo, DP; Flanigan, MJ; Kravitz, SA; Mobarry, CM؛ و دیگران ((March 2000)). "A whole-genome assembly of Drosophila". Science. 287 (5461): 2196–204. صص. http://science٫sciencemag٫org/content/۲۸۷/۵۴۶۱/۲۱۹۶. تاریخ وارد شده در
|سال=را بررسی کنید (کمک) - Batzoglou, S. ; Jaffe, DB; Stanley, K; Butler, J; Gnerre, S; Mauceli, E; Berger, B; Mesirov, JP; Lander, ES ((January 2002)). "ARACHNE: a whole-genome shotgun assembler". Genome Research. 12 (1): 177–89. صص. http://genome٫cshlp٫org/content/۱۲/۱/۱۷۷٫long. تاریخ وارد شده در
|سال=را بررسی کنید (کمک) - «AMOS».
- «Pyrosequencing - Wikipedia».
- Dohm, J. C. ; Lottaz, C. ; Borodina, T. ; Himmelbauer, H. ((November 2007)). "SHARCGS, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing". Genome Research. 17 (11): 1697–706. صص. http://www٫genome٫org/cgi/pmidlookup?view=long&pmid=۱۷۹۰۸۸۲۳. تاریخ وارد شده در
|سال=را بررسی کنید (کمک) - «De-Novo Transcription Assembly - Wikipedia».
- Kamath, Govinda M.; Shomorony, Ilan; Xia, Fei; Courtade, Thomas; Tse, David N. (1 August 2016). "HINGE: Long-Read Assembly Achieves Optimal Repeat Resolution" (PDF). biorXiv preprint.
- Grabherr, Manfred G.; Haas, Brian J.; Yassour, Moran; Levin, Joshua Z.; Thompson, Dawn A.; Amit, Ido; Adiconis, Xian; Fan, Lin; Raychowdhury, Raktima (2011-07-01). "Full-length transcriptome assembly from RNA-Seq data without a reference genome". Nature Biotechnology. 29 (7): 644–652. doi:10.1038/nbt.1883. ISSN 1087-0156. PMC 3571712. PMID 21572440.
- Boisvert, Sébastien; Laviolette, François; Corbeil, Jacques (October 2010). "Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies". Journal of Computational Biology. 17 (11): 1519–33. doi:10.1089/cmb.2009.0238. PMC 3119603. PMID 20958248.