بلاست
بلاست (به انگلیسی: BLAST) نام یک نرمافزار کاربردی در علوم سلولی و مولکولی و ژنتیک است که مخفف واژگان Basic Local Alignment Search Tool یا ابزار پایهای برای جستجوی برهمنهیهای موضعی است.[1] این ابزار قسمتی از مجموعهٔ اطلاعات کیفی مرکز ملی اطلاعات زیستفناوری است.[2]

با این نرمافزار میتوان توالی اسیدهای آمینه در پروتئینها یا توالی نوکلئوتیدها را در DNA را با هم مقایسه کرد. این نرمافزار به پژوهشگر اجازه میدهد تا یک توالی را با توالی دیگر یا توالی که در بانک اطلاعاتی وجود دارد، مقایسه کند. شناسایی توالیهای موجود در بانک اطلاعاتی که بیشترین شباهت را با توالی مورد نظر دارد از دیگر قابلیتهای این نرمافزار است. بر حسب نوع توالی انواع مختلفی از بلاست امکانپذیر است. مثلاً اگر یک ژن ناشناخته در موش که قبلاً اطلاعاتی از آن در اختیار نبوده، باید بررسی شود، یک پژوهشگر ترجیح میدهد این توالی را با ژنوم انسان بلاست کند. این نرمافزار در NIH (مؤسسه ملی بهداشت آمریکا) طراحی شد. بلاست یکی از پرکاربردترین نرمافزارها در بیوانفورماتیک است که با سرعت مطلوب مقایسه مورد نظر را انجام میدهد. سرعت زمانی اهمیت خود را نشان میدهد که با ژنوم کامل روبرو باشیم. پیش از طراحی این نرمافزار مقایسه توالیها بسیار وقت گیر بود.
پیشینه
به دلیل اینکه بلست مربوط به یکی از بنیادیترین مسائل بیوانفورماتیک است و سرعت خوبی دارد یکی از پر کاربردترین نرمافزارهای بیوانفورماتیک به حساب میآید. سرعت، مخصوصاً در کاربردهای واقعی روی پایگاههای دادهٔ بزرگ ژنوم امری حیاتی است.
پیش از ارائه الگوریتمهای سریعی مثل BLAST و FASTA جستجو در پایگاههای دادهٔ بزرگ برای دنبالههای پروتئین یا نوکلئیک با استفاده از الگوریتمهای انطباق کامل مثل Smith-Waterman بسیار زمانگیر بود. BLAST از نظر زمانی کارآمد تر از FASTA است زیرا الگوهای مهمتر در دنباله را مورد جستجو قرار میدهد.
از BLAST در موارد زیر استفاده میشود:
- کدام گونه باکتریها پروتئینی دارند که از نظر اصل و نسب به پروتئینی با آمینواسید شناخته شده مرتبط باشد؟
- دنبالهٔ معلومی از DNAها از کجا سرچشمه گرفتهاند؟
- کدام ژنهای دیگر پروتئینهایی را به وجود میآورند که ساختارشان با ساختارهایی که قبلاً مشخص شده است یکی باشد؟
الگوریتم بلاست و برنامهٔ کامپیوتری آن توسط Stephen Altschul, Warren Gish و David Lipman در مؤسسه ملی اطلاعات بیوتکنولوژی آمریکا(NCBI) (شاخهای از NIH)و Web Myers در ایالت پنسیلوانیا و Gen Myers در دانشگاه Arizona پیادهسازی شد.
ورودی
دنبالههای ورودی در فرمت FASTA format یا Genbank هستند.
خروجی
خروجی بلاست در فرمتهای مختلفی موجود است: فرمت HTML، متن و XML. در نرمافزار موجود در سایت NCBI فرمت پیشفرض خروجی HTML است. در این نرمافزار موفقیتها (مواردی که تطابق رخ داده) به صورت گرافیکی نمایش داده میشوند بعلاوهٔ یک جدول که دنبالهٔ تطابقها همراه با امتیاز داده شده به هرکدام را نشان میدهد.
همچنین یک نسخهٔ رایگان بلاست قبل اجرا بر روی کامپیوتر شخصی از آدرس BLAST + Exe قبل دانلود است. پایگاه داده نیز از سایت NCBI و هم چنین از آدرس پایگاه داده بلاست قابل دانلود است.
نحوه پردازش
با استفاده از روش Heuristic الگوریتم بلاست دنبالههای همسان را پیدا میکند. در این روش به جای مقایسه کامل دو دنباله، تطابقهای کوتاه با یکدیگر مقایسه میشوند. به پروسهٔ پیدا کردن کلمات اولیه برای اجرای الگوریتم بلاست seeding میگویند. بعد از پیدا کردن این تطابقهای اولیه الگوریتم بلاست یک تطابق محلی (local alignment) انجام میدهد. هنگام پیدا کردن هومولوگ در دنبالهها، مجموعه حروف مشترک که به آنها لغت گفته میشود بسیار مهم اند. به عنوان مثال اگر دنبالهای شامل حروف GLKFA باشد؛ اگر بلاست با تنظیمات پیشفرض اجرا شود، طول لغت ۳ خواهد بود(word size = 3). در این حالت لغاتی که جستجو خواهند شد لغات روبه رو هستند: GLK, LKF, KFA. الگوریتم اکتشافی (heuristic) بلاست، حروف سه تایی مشترک را بین دنبالهٔ مورد نظر و دنبالهٔ تطابق یا دنبالههای پایگاه داده مکان یابی میکند. سپس از این نتایج برای ساخت یک تطابق استفاده میشو. بعد از ساخت لغات برای دنبالهٔ مورد نظر، لغات موجود در همسایگی نیز ساخته میشوند. این لغات باید در پروسهٔ امتیاز دهی، امتیازشان از یک حد آستانهای بیشتر شده باشد؛ و معمولاً برای این امتیاز دهی از ماتریس BLOSUM62 استفاده میشود. بعد از ساخت لغات و برای پیدا کردن تطابق لفات ساخته شده با دنبالههای موجود در پایگاه داده مقایسه میشوند. حد آستانه مشخص میکند که لغت مورد نظر در تطابق نهایی باشد یا نباشد. بعد از انجام پروسهٔ seeding (پیدا کردن تطابق اولیه)، تطابق یافته شدهٔ اولیه که در این مثال طول آن سه بود در دو جهت گسترش مییابد و با هر گسترش امتیاز جدیدی به تطابق داده میشود و اگر این امتیاز از مقداری از قبل تعیین شده بیشتر بود تطابق پذیرفته میشود و در غیر اینصورت از گسترش تطابق خودداری میشود. افزایش مقدار از قبل تعیین شده، فضای جستجو را محدود میکند و تعداد لغات همسایگی را کاهش میدهد اما سرعت اجرای الگوریتم بلاست را افزایش میدهد.
الگوریتم
برای اجرای بلاست نیاز به دو دنباله میباشد یکی دنبالهٔ درخواستی یا مورد نظر و دیگری دنبالهٔ هدف یا دنبالههای موجود در پایگاه دادهای از دنبالهها. بلاست زیر دنبالههایی از پایگاه داده را پیدا میکند که شبیه دنبالهٔ ورودی (query) باشند. معمولاً دنبالهٔ query بسیار کوچکتر از پایگاه داده است. به عنوان مثال query ممکن است هزار نوکلئوتیدی باشد در حالی که پایگاه داده از چندین بیلیون نوکلئوتید تشکیل شده باشد. ایدهٔ اصلی الگوریتم بلاست این است که به دنبال تطابقهای با بیشترین امتیاز بین query و پایگاه داده میگردد بر اساس تقریب زدن یک الگوریتم اکتشافی به اسم Smith-Waterman_algorithm. الگوریتم Smith-Waterman بسیار زمانگیر است از اینرو در بلاست از یک روش اکتشافی (heuristic) که البته دقت کمتری خواهد داشت استفاده میشود. اما در عوض ۵۰ بار سریعتر است. در زیر مراحل الگوریتم بلاست (پروتئین به پروتئین) به صورت خلاصه آورده شده است:
- حذف قسمتهای تکراری از دنبالهٔ query (قسمتهای با پیچیدگی کم): قسمتهای با پیچیدگی کم قسمتهایی در دنباله هستند که عناصرشان تنوع کمی دارند.
- ساخت یک لیست k حرفی از دنباله query:
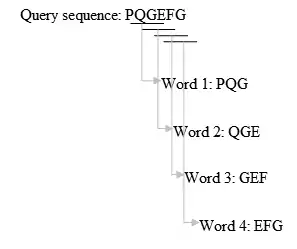
- لیست کردن تطابقهای ممکن:
این مرحله یکی از تفاوتهای اساسی بین بلاست و FASTA است. برای FASTA تمام لغات مشترک در پایگاه داده و دنبالههای کوئری ای که در مرحلهٔ دوم لیست شدند مهم است، اما برای بلاست فقط لغات با امتیاز بالا اهمیت دارند.
- سازماندهی لغات باقیمانده (لغات با امتیاز بیشتر از حد آستانه) و تبدیل آن به یک درخت جستجوی بهینه:
 هدف این قسمت این است که برنامه بتواند سریعتر لغات با امتیاز بالا را با دنبالههای پایگاه داده مقایسه کند.
هدف این قسمت این است که برنامه بتواند سریعتر لغات با امتیاز بالا را با دنبالههای پایگاه داده مقایسه کند.
- تکرار مراحل ۳ و ۴ به ازای هر لغت k-حرفی در دنباله query.
- جستجو در پایگاه داده برای پیدا کردن تطابق دقیق بین دنبالههای باقیمانده و دنبالههای پایگاه داده:
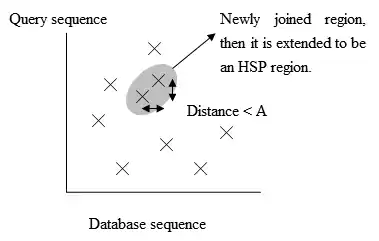
- گسترش تطابقهای دقیق به HSP (جفت دنبالههای با امتیاز تطابق بالاتر از حد آستانه)
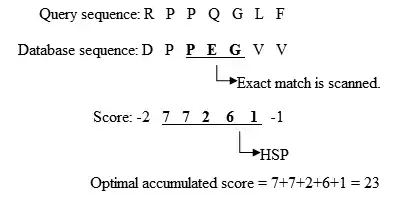
- بررسی معناداری امتیاز داده شده به HSPها.
در این مرحله، بلاست میزان معناداری آماری امتیاز داده شده به هر HSP را با استفاده از Gumbel extreme value distribution (EVD) بررسی میکند. بر طبق Gumbel EVD احتمال اینکه با احتمال p امتیاز مشاهده شود مثل S که بزرگتر یا مساوی x باشد برابر است با:

به طوریکه:

- ادغام HSPها و تبدیل آنها به تطابقهای بزرگتر
- نمایش تطابق محلی دنباله query با پایگاه داده با استفاده از الگوریتم gapped Smith-Waterman.
- انتخاب تطابقهایی که مقدار مورد انتظار امتیاز آنها کمتر از حد آستانهٔ E شده باشد.
بلاست موازی
نسخههای موازی از بلاست با استفاده از MPI و Pthreads پیادهسازی شدهاند و روی پلتفرمهای مختلف مثل ویندوز، لینوکس، مک و… قابل اجرا هستند. روشهای مشهور برای موازی سازی بلاست عبارتند از پراکنده یا پخش کردن query، قطعه قطعه سازی جدول hash، موازی سازی محاسباتی و قطعه قطعه سازی پایگاه داده.
برنامه
بلاست هم میتواند دانلود شده و به صورت command-line اجرا شود (برنامه blastall)و هم اینکه بدون دانلود روی وب اجرا شود. بلاست یک برنامه متن باز است و هرکس میتواند در کد تغییرات مورد نظر خود را اجرا کند و این خود علت وجود نسخههای مختلف از بلاست میباشد. بلاست مجموعهای از برنامههای مختلف است که همگی در فایل اجرایی blastall قابل دسترسی است. ایم مجموعهها شامل:
- نوکلئوتید-نوکلئوتید بلاست (blastn)
- پروتئین-پروتئین بلاست (blastp)
- بلاست تکرار شوندهٔ وابسته به مکان (position-specific iterative blast)
در این روش ابتدا یک پروفایل عمومی ساخته میشود. سپس با استفاده از این پروفایل گروههای بیشتری از پروتئینها به دست میآیند که خود آنها نیز تشکیل یک پروفایل جدید میدهند و این کار تکرار میشود.
- Nucleotide 6-frame translation-protein
این برنامه به صورت ۶-فریم ۶-فریم ترجمهٔ دنبالهٔ query را با دنباله پروتئین مقایسه میکند.
- Nucleotide 6-frame translation-nucleotide 6-frame translation (tblastx)
این برنامه مانند قبلی است با این تفاوت که هر دو را ترجمه میکند و ترجمهها را با هم مقایسه میکند.
- Protein-nucleotide 6-frame translation (tblastn)
این برنامه query پروتئین را با ترجمه نوکلئوتید مقایسه میکند.
- Large numbers of query sequences (megablast)
این برنامه هنگام مقایسهٔ دنبالههای بزرگ استفاده میشود.
نسخههای جایگزین
یکی از نسخههای بلاست که خیلی سریع تر است اما دقت کمتری دارد BLAT است. نسخهٔ دیگری که برای مقایسهٔ ژنومها و کروموزومهای خیلی بزرگ استفاده میشود BLASTZ نام دارد. CS-BLAST (context-specific BLAST) نیز یک نسخهٔ توسعه یافتهٔ بلاست برای جستجو در پروتئین هاست که دو برابر سریعتر از بلاست است. در این نسخه احتمال جهش بین آمینواسیدها نه تنها به خودشان بلکه به مکان آنها نیز وابسته است.
نسخههای سریعتر
- دو پیادهسازی اصلی در این رده progeniq و Tera-BLAST هستند که به عنوان مثال progeniq صد برابر سریعتر از بلاست معمولی است.
- Mitrion-C Open نیز نسخهٔ دیگری است که از لینک http://mitc-openbio.sourceforge.net/ قابل دسترسی است.
- نسخهٔ دیگری از بلاست که GPU-based است از CUDA-BLASTP قابل دسترسی است که ده برابر سریعتر از NCBI BLAST است.
کاربردهای بلاست
از بلاست میتوان در مواردی مثل موارد زیر استفاده نمود:
- تعیین نوع
با استفاده از بلاست میتوان هومولوگهای یک نوع را پیدا کرد.
- قرار دادن دامنهها
هنگام کار با پروتئینها میتوان آنها را به عنوان ورودی به بلاست داد تا دامنههای شناخته شده از دنبالهٔ مورد نظر را پیدا کند.
- کشیدن درخت فیلوژنتیک
با استفاده از نتایج به دست آمده از بلاست میتوان درخت فیلوژنتیک را رسم کرد.
- نگاشت DNA
هنگام جستجو به دنبال دنبالهای از ژنها در مکانی ناشناخته روی یک گونهٔ شناخته شده، بلاست میتواند مکانهای کروموزومی در دنبالههای مورد نظر مقایسه کند.
- مقایسه
هنگام کار کردن با ژنها بلاست میتواند ژنهای مشترک در دو گونهٔ متفاوت را شناسایی کند.
مقایسهٔ BLAST و Smith-Waterman
اگرچه blast و Smith-Waterman هردو برای یافتن هومولوگها استفاده میشوند اما تفاوتهایی نیز با یکدیگر دارند. بلاست به خاطر اینکه یک الگوریتم اکتشافی (heuristic) است دقت کم تری دارد و ممکن است بعضی از تطابقها را پیدا نکند و از دست بدهد اما در عوض سرعت خوبی دارد. در مقابل Smith-Waterman دقت مناسبی دارد اما در مقایسه با بلاست زمانگیرتر است و فضای حافظهٔ بیشتری نیاز دارد؛ اما در عوض برای یافتن همومولوگهای دور مفید است. خوشبختانه تکنولوژیهایی برای سرعت بخشیدن به Smith-Waterman ارائه شدهاند که به عنوان مثال میتوان استفاده از چیپهای FPGA یا پردازشهای SIMDرا نام برد. برای گرفتن نتایج بهتر از بلاست میتوان تنظیمات مختلفی که برای این برنامه وجود دارد را تغییر داد، اما روشی وجود ندارد که برای هر دنبالهٔ دلخواه بهترین تنظیمات را ارائه کند. این تنظیمات شامل E-value , gap costs, filters, word size, وsubstitution matrix میباشند.
پیوند به بیرون
منابع
- Altschul, Stephen F. , Warren Gish, Webb Miller, Eugene W. Myers, and David J. Lipman (1990). Basic local alignment search tool. J. Mol. Biol. 215:403-10
- Bioinformatics basics: applications in biological science and medicine. Hooman H. Rashidi, Lukas K. Buehler. Publisher CRC Press, 2000. ISBN 0-8493-2375-4 pp.39
- مشارکتکنندگان ویکیپدیا. «BLAST». در دانشنامهٔ ویکیپدیای انگلیسی، بازبینیشده در ۳ می ۲۰۱۱.
| پایگاههای داده |
|
|---|---|
| نرمافزارها | |
| سایر |
|
| مؤسسات |
|
| سازمانها |
|
| اجلاسها |
|
| فرمتهای فایلی | |
| موضوعات مرتبط |
|