کمبود لیپوپروتئین لیپاز
کمبود لیپوپروتئین لیپاز (انگلیسی: Lipoprotein lipase deficiency) که با عناوین دیگری نظیر «سندرم شیلومیکرونمی فامیلیال»،[1] «شیلومیکرونمی»[2]، «سندرم شیلومیکرونمی»[3] و «هایپرلیپوپروتئینمی نوع Ia»[4] هم شناخته میشود، یک بیماری نادر ارثی است که در اثر جهش در ژن کدکنندهٔ آنزیم لیپوپروتئین لیپاز ایجاد میشود.[2] در نتیجه، افراد مبتلا دچار فقدان این آنزیم مهم بوده و قادر به هضم و تجزیهٔ تریگلیسریدها نیستند.
| کمبود لیپوپروتئین لیپاز | |
|---|---|
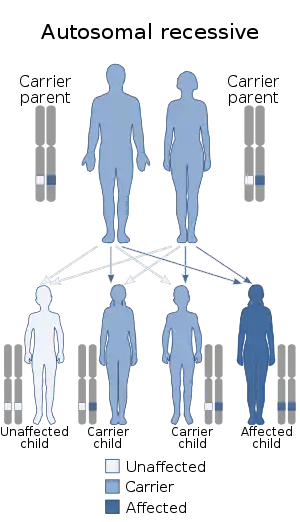 | |
| توارث این بیماری از نوع اتوزومال مغلوب است. | |
| طبقهبندی و منابع بیرونی | |
| تخصص | غدد درون ریز و متابولیسم |
| آیسیدی-۱۰ | E78 |
| اُمیم | ۲۳۸۶۰۰ |
| دادگان بیماریها | 4697 |
| مدلاین پلاس | 000408 |
| سمپ | D008072 |
| مرور ژن | |
| اورفانت | ۴۴۴۴۹۰ |
در پلاسمای این افراد سطح شیلومیکرونها بشدت بالا میرود و هیپرتریگلیسریدمی رخ میدهد. (معمولاً سطح ناشتای تریگلیسریدهای خون در این بیمارن بالای ۲۰۰۰ میلیگرم در دسیلیتر است) علائم این بیماری در دوران نوزادی، دردهای کولیکی شکم، اختلال رشد است. در تمامی سنین، شایعترین علامت بیماری، دردهای راجعهٔ شکمی و پانکراتیت حاد است. این درد ممکن است در بالای شکم باشد و به پشت بزند، یا آنکه در تمامی شکم گسترده باشد و علائم یک شکم حاد اورژانسی را تقلید کند. از سایر علائم میتوان به بزرگشدن کبد و طحال (هپاتواسپلنومگالی)، زانتومای فورانی (تقریباً در ۵۰٪ بیماران) و تجمع چربی در شبکیه (لیپمیا رتینالیس) اشاره کرد. بروز پانکراتیتهای حاد، علاوه بر خطر جانی، میتواند احتمال نارسایی مزمن لوزالمعده و دیابت را بدنبال داشته باشد.
میزانِ بروز این بیماری، ۱ در یک میلیون نفر است.[5]
جستارهای وابسته
منابع
- Santamarina-Fojo, S (1998). "Familial lipoprotein lipase deficiency". Endocrinol Metab Clin North Am. 27 (3): 551–567. PMID 9785052.
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0. OCLC 62736861.
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0. OCLC 212399895.
- Online 'Mendelian Inheritance in Man' (OMIM) HYPERLIPOPROTEINEMIA, TYPE I -238600, updated 03/18/2004. As of October 2012, mention of type Ia no longer appears in the OMIM record.
- A.D.A.M. Editorial Board (2011-05-29). Dugdale, III, David C.; Zieve, David, eds. Familial lipoprotein lipase deficiency. A.D.A.M. Medical Encyclopedia. National Center for Biotechnology Information (published May 29, 2011). Retrieved October 15, 2012
- مشارکتکنندگان ویکیپدیا. «Lipoprotein lipase deficiency». در دانشنامهٔ ویکیپدیای انگلیسی، بازبینیشده در ۸ اکتبر ۲۰۱۷.