بلوسام
ماتریسهای بلوسام (BLOSUM) (ماتریس بلوکهای جایگزینی) یک ماتریس جایگزینی است که در هم ترازی توالیهای پروتئینی استفاده میشود. این ماتریسها برای امتیازدهی هم ترازیهای توالیهای پروتئینی که در تکامل از یکدیگر متمایز شدهاند، استفاده میشود و بر مبنای هم ترازی محلی میباشند. ماتریس بلوسام برای اولین بر در مقالهای توسط Henikoff معرفی شد.[1] آنها پایگاه دادهٔ بلوکها را برای پیدا کردن مناطق حفاظت شده در پروتئینها پیمایش کردند (مناطقی که در هم ترازی دنباله وقفه وجود ندارد) و فرکانسهای آمینو اسیدهای مرتبط و احتمالهای جایگزینی را به دست آوردند. سپس امتیاز لوجیت هر ۲۱۰ جایگزینی ممکن بین ۲۰ پروتئین استاندارد را محاسبه نمودند. بر خلاف ماتریسهای PAM که بر پایه مقایسه بین پروتئین نزدیک برونیابی میشود، ماتریسهای بلوسام بر پایهٔ هم ترازیهای مشاهدهشده ساخته شدهاست.
پس زمینهٔ زیستی
دستورالعملهای ژنتیکی هر سلول از یک موجود زنده در DNA آن ذخیره شده است.[2] در طول حیات سلول، این اطلاعات برای تولید پروتئین یا برای تقسیم سلولی رونویسی میشوند و احتمال آن وجود دارد که این محتویات در حین این فرایندها دستخوش تغییر شوند.[2][3] این تغییر به عنوان جهش شناخته شده است. در سطح مولکولی سیستمهای تنظیم کننده ای هستند که بیشتر این جهشها را اصلاح میکنند.[3][4]
عملکرد پروتئینها بسیار وابسته به ساختار آنها است.[5] تغییر یک آمینواسید در پروتئین ممکن است کارآمدی آن را برای انجام وظیفهٔ مربوطه کاهش یا کارکرد آن را تغییر دهد.[3] تغییرات این چنینی میتوانند یک عملکرد حیاتی در سلول را مختل کنند یا حتی منجر به مرگ سلول شوند.[6] در مقابل، این تغییر ممکن است به سلول اجازه ادامهٔ فعالیت هرچند متفاوت را بدهدو جهش به فرزندان موجودات زنده منتقل شود. اگر این تغییر باعث ضعف جسمی قابل توجهی نشود این احتمال وجود دارد که جهش در جمعیت باقی بماند. همچنین این امکان وجود دارد که تغییر در عملکرد یک تغییر مفید باشد.
۲۰ اسید آمینه ترجمه شده توسط کد ژنتیکی تا حدود زیادی از نظر خواص فیزیکی و شیمیایی زنجیرههای جانبی آنها متفاوت هستند.[5] این اسیدهای آمینه میتواند به طبقهبندی به گروههای مشابه با خواص فیزیکوشیمیایی.[5] جایگزین کردن یک اسید آمینه با یکی دیگر از همان دسته است بیشتر احتمال دارد به یک کوچکتر تأثیر بر ساختار و عملکرد پروتئین از جایگزینی با یک اسید آمینه از ردههای مختلف.
همترازی توالیها یک روش اساسی برای تحقیقات زیستشناسی مدرن است. رایجترین همردیفی توالیهای پروتئین، جستجوی شباهت بین توالیهای مختلف به منظور فهم تفاوت تکاملی توالیهای پروتئینی به منظور پیش بینی وظیفهٔ ژنهای جهش یافته است. ماتریسها در الگوریتمهای محاسبهٔ میزان شباهت توالیها استفاده میشوند[1]
اصطلاحات
بلوسام: ماتریس جایگزینی بلوکها، یک ماتریس جایگزینی که برای هم ترازی توالیهای پروتئین استفاده میشود.
ماتریسهای امتیازدهی (آمار در مقابل زیستشناسی): برای معنا یافتن ارزیابی هم ترازی توالیها نیاز به ماتریس امتیازدهی یا جدولی است که بیانگر احتمال جایگزینی معنادار جفت آمینواسیدها یا جفت نوکلئوتیدها در یک همتراری است. امتیازها برای هر موقعیت مکانی از همترازی محلی پروتئینها بدست میآیند.[7]
دستههای متعددی از ماتریسهای بلوسام با استفاده از پایگاههای داده وابسته به هم ترازیهای متفاوت وجود دارند که با عددهای متفاوت نامگذاری میشوند. ماتریسهای بلوسام با اعداد بزرگتر برای مقایسهٔ توالیهای نزدیک به هم طراحی شدهاند در حالیکه ماتریسهایی با اعداد کوچکتر این عمل را برای توالیهای نسبتاً دور انجام میدهند. به عنوان مثال BLOSUM80 برای هم ترازی توالیهایی با تفاوت کمتر و BLOSUM45 برای هم تراری توالیهای متفاوت تر استفاده میشود
ماتریسها توسط ادغام کردن همهٔ دنبالههایی که از درصدی که به یک دنباله داده میشود شبیه تر هستند ساخته میشود، و سپس تنها آن دنبالهها را مقایسه میکند. درصد مذکور به نام ماتریس افزوده میشود. برای نمونه BLOSUM۸۰، از ادغام کردن دنبالههایی با یکسانی بیش از ۸۰ درصد تولید میشود.[1]
ساخت ماتریسهای بلوسام
ماتریسهای بلوسام با استفاده از اعمال روشهای آماری بر بلوکهای آمینواسیدهای مشابه برای بدست آوردن امتیازهای شباهت بدست میآیند.
مراحل روشهای آماری:[8]
حذف توالی
حذف توالیهای با میزان شباهت بیشتر از r%.
دو روش برای حذف توالیها وجود دارد. یا میتوان توالیها را از بلوک مربوطه حذف کرد یا توالیهای مشابه را یافته و با توالیهای جدیدی که میتوانید نمایندهٔ خوشههای مربوطه باشند جایگزین نمود. این عمل برای جلوگیری از بایاس نتیجه به نفع پروتئینهای مشابه صورت میگیرد.
محاسبه فرکانس و احتمال
پایگاه داده ای برای ذخیرهسازی هم ترازی توالیهایی از نواحی با بیشترین حفاظت از خانوادهٔ پروتئینها.
این هم ترازیها برای بدست آوردن ماتریس بلوسام استفاده میشوند. نواحی حفاظت شده، نواحی ای از آمینواسیدها هستند که تغییر جزئی بین آنها وجود دارد.
نرخ Log Odd
از رابطهٔ زیر بدست میآید.

که در آن احتمال مشاهده شده و احتمال مورد انتظار است.


BLOSUM ماتریس
میزان شانس شباهت توسط نرخ Log Odd محاسبه شده و ماتریسهای بلوسام از گرد کردن این مقادیر بدست میآیند.
امتیاز ماتریسهای بلوسام
یک ماتریس امتیازدهی یا جدولی از مقادیر برای ارزیابی اهمیت هم ترازی توالیها مورد نیاز است. به طور کلی وقتی دو توالی نوکلئوتیدی مقایسه می شوندتمامی آنچه در امتیازدهی در نظرگرفته میشود آن است که دو باز در مکانهای متناظر یکسان هستند یا نه. تمامی برابریها و عدم برابریها امتیاز یکسانی دارند.[9] ولی این قضیه در رابطه با پروتئینها متفاوت است و ماتریسهای جایگزینی برای آمینواسیدها پیچیدهتر هستند و تمامی عواملی که ممکن است فرکانس جایگزینی را تغییر دهد در نظر گرفته میشوند که در نتیجهٔ آن پنالتی نسبتاً زیاد برای همترازیهایی است که احتمال همولوگ بودن آنها پایین است.[7]
ماتریسهای جایگزینی ای که به صورت عمده استفاده میشوند ماتریسهای بلوسام (BLOSUM)[1] و ماتریسهای جهش نقطه ای پذیرفته شده (PAM)[10][11] هستند. این دو ماتریس با روشهای متفاوتی محاسبه میشوند.[7]
امتیازات در بلوسام امتیازات log_odds هستند که در یک همترازی از نسبت درستنمایی دو آمینو اسید که بیولوژیکی ظاهر شدهاند به درستنمایی هر یک از دو آمینو اسیدی که به صورت اتفاقی ظاهر شدهاند، محاسبه میشود. مقدار مثبت، محتمل تر بودن جایگزینی و امتیاز منفی، غیر محتمل بودن جایگزینی را نتیجه میدهد.[12][13]
تساوی زیر برای محاسبه ماتریس BLOSUM استفاده میشود:

در اینجا احتمال جابجا شدن دو آمینو اسید و در دنبالههای مشابه (هومولوگ) و و احتمال رخ دادن آمینو اسید و به صورت تصادفی در دنباله پروتیینها میباشد. به منظور اینکه ماتریس حاوی مقادیر صحیح ساده باشد از فاکتور استفاده میکنیم.






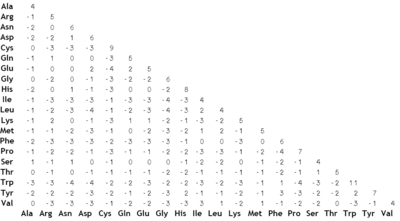
مثال - BLOSUM62
بلوسام۶۲: پروتئینهای با ارتباط متوسط
بلوسام۸۰: پروتئینهای مرتبط تر
بلوسام۴۵: پروتئینهای با ارتباط کمتر
مقالهای در Nature Biotechnology[14] نشان داد کهBLOSUM۶۲ که سالیان سال است به عنوان استاندارد استفاده میشود طبق الگوریتمی که هنیکوف ارائه داده دقیقاً صحیح نیست.[1] در کمال تعجب، بلوسام اشتباه محاسبه شده، کارایی جستجو را ارتقا میدهد.[14]
برخی از کاربردهای ماتریس بلوسام در بیوانفورماتیک
کاربردهای پژوهشی
امتیازهای بلوسام در پیش بینی و درک انواع ژن سطحی در میان حاملهای ویروس هپاتیت B حامل[15] و اپیتوپهای لنفوسیت تی مورد استفاده قرار گرفته است[16]
استفاده در BLAST
ماتریسهای بلوسام هم چنین به عنوان ماتریس امتیاز دهی در مقایسهٔ توالیهای DNA و توالیهای پروتئین برای ارزیابی کیفیت هم ترازی استفاده میشود. برای این شکل از سیستم امتیازدهی نرمافزارهای گسترده ای من جمله بلاست وجود دارد.[17]
مقایسه PAM و BLOSUM
علاوه بر ماتریسهای BLOSUM ماتریسهای قبلاً توسعه یافته PAM میتوانند استفاده شوند.[1]
از آنجا که هر دو ماتریس PAM و BLOSUM روشهای متفاوتی برای نمایش اطلاعات امتیازدهی یکسانی هستند میتوان این دو را مقایسه نمود اما به دلیل تفاوت زیاد روش بدست آوردن این اطلاعات BLOSUM100 با PAM100 یکی نیست.[18]
| PAM | BLOSUM |
|---|---|
| PAM100 | BLOSUM90 |
| PAM120 | BLOSUM80 |
| PAM160 | BLOSUM60 |
| PAM200 | BLOSUM52 |
| PAM250 | BLOSUM45 |
رابطه بین PAM و BLOSUM
| PAM | BLOSUM |
|---|---|
| برای مقایسهٔ توالیهای بسیار مرتبط،
ماتریسهای PAM با اعداد کمتر استفاده میشوند |
برای مقایسهٔ توالیهای بسیار مرتبط،
ماتریسهای BLOSUM با اعداد بزرگتر استفاده میشوند |
| برای مقایسهٔ توالیهای با ارتباط کمتر
ماتریسهای PAM با اعدادبزرگتر استفاده میشوند |
برای مقایسهٔ توالیهای با ارتباط کمتر
ماتریسهای PAM با اعداد کوچکتر استفاده میشوند |
تفاوت بین PAM و BLOSUM
| PAM | BLOSUM |
|---|---|
| بر مبنای هم ترازی گلوبال
توالیهای نزدیک هم است |
بر مبنای هم ترازی محلی است |
| PAM1 ماتریسی است که از مقایسهٔ توالیهایی
بدست میآید که کمتر از ۱٪ اختلاف دارند. |
BLOSUM62 ماتریسی است که از مقایسهٔ توالیهایی بدست میآید
که کمتر از ۶۲٪ به هم شبیه هستند |
| سایر ماتریسهای PAM از برون یابی ماتریس PAM1 بدست آمدهاند. | بر مبنای هم ترازیهای مشاهده شده است
و از توالیهای پروتئین نزدیک برون یابی نشده است. |
| عددهای بزرگتر در نام گذاری این ماتریسها بیانگر فاصلهٔ تکاملی بیشتر است. | عددهای بزرگتر در نام گذاری این ماتریسها بیانگر
میزان شباهت بیشتر در توالیها و در نتیجه فاصلهٔ تکاملی کمتر است.[19] |
جستارهای وابسته
منابع
- Henikoff, S.; Henikoff, J.G. (1992). "Amino Acid Substitution Matrices from Protein Blocks". PNAS. 89 (22): 10915–10919. doi:10.1073/pnas.89.22.10915. PMC 50453. PMID 1438297.
- Campbell NA; Reece JB; Meyers N; Urry LA; Cain ML; Wasserman SA; Minorsky PV; Jackson RB (2009). Biology: Australian Version (8th ed.). Pearson Education Australia. pp. 307–325. ISBN 978-1-4425-0221-5.
- Campbell NA; Reece JB; Meyers N; Urry LA; Cain ML; Wasserman SA; Minorsky PV; Jackson RB (2009). Biology: Australian Version (8th ed.). Pearson Education Australia. pp. 327–350. ISBN 978-1-4425-0221-5.
- Fundamentals of Molecular Biology (1st ed.). Oxford University Press. 2009. pp. 187–203. ISBN 978-0-19-569781-0.
- Campbell NA; Reece JB; Meyers N; Urry LA; Cain ML; Wasserman SA; Minorsky PV; Jackson RB (2009). Biology: Australian Version (8th ed.). Pearson Education Australia. pp. 68–89. ISBN 978-1-4425-0221-5.
- Lobo, Ingrid (2008). "Mendelian Ratios and Lethal Genes". Nature Publishing Group. Retrieved 19 October 2013.
- pertsemlidis A.; Fondon JW.3rd (September 2001). "Having a BLAST with bioinformatics (and avoiding BLASTphemy)". genome biology. 2 (10): reviews2002.1–2002.10. PMID 11597340.
- "BLOSSUM MATRICES: Introduction to BIOINFORMATICS" (PDF). UNIVERSITI TEKNOLOGI MALAYSIA. 2009. Retrieved 9 September 2014.
- Murali Sivaramakrishnan; Ognjen Perisic; Shashi Ranjan. "CS#594 - Group 13 (Tools and softwares)" (PDF). University of Illinois at Chicago - UIC. Retrieved 9 September 2014.
- Margaret O., Dayhoff (1978). "22". Atlas of Protein Sequence and Structure. 5. Washington DC: National Biomedical Research Foundation. pp. 345–352.
- States DJ.; Gish W.; Altschul SF. (1991). "Improved sensitivity of nucleic acid database searches using application-specific scoring matrices". Methods: A Companion to Methods in Enzymology. 3: 66–70. doi:10.1016/s1046-2023(05)80165-3. ISSN 1046-2023.
- Albert Y. Zomaya (2006). Handbook of Nature-Inspired And Innovative Computing. New York, NY: Springer. ISBN 0-387-40532-1.
- NIH "Scoring Systems"
- Mark P Styczynski; Kyle L Jensen; Isidore Rigoutsos; Gregory Stephanopoulos (2008). "BLOSUM62 miscalculations improve search performance". Nat. Biotechnol. 26 (3): 274–275. doi:10.1038/nbt0308-274. PMID 18327232.
- "Viral and clinical factors associated with surface gene variants among hepatitis B virus carriers". Antivir Ther. 12 (8): 1255–1263. 2007. PMID 18240865.
- "Reliable prediction of T‐cell epitopes using neural networks with novel sequence representations" (PDF). Protein Science. 12 (5): 1007–1017. 2003. doi:10.1110/ps.0239403.
- "The Statistics of Sequence Similarity Scores". National Centre for Biotechnology Information. Retrieved 20 October 2013.
- Saud, Omama (2009). "PAM and BLOSSUM SUBSITUTION MATRICES". Birec. Archived from the original on 9 March 2013. Retrieved 20 October 2013.
- "The art of aligning protein sequences Part 1 Matrices". Dai hoc Can Tho - Can Tho University. Archived from the original on 11 September 2014. Retrieved 7 September 2014.
پیوند به بیرون
- Sean R. Eddy (2004). "Where did the BLOSUM62 alignment score matrix come from?". Nature Biotechnology. 22 (8): 1035–6. doi:10.1038/nbt0804-1035. PMID 15286655.
- بلوک WWW سرور
- سیستم امتیاز دهی برای انفجار در NCBI
- فایلهای داده از BLOSUM در NCBI سرور FTP.
- تعاملی BLOSUM شبکه تجسم بایگانیشده در ۳۰ ژانویه ۲۰۱۷ توسط Wayback Machine