سندرم دوبوویتز
سندرم دوبوویتز (انگلیسی: Dubowitz syndrome) یک اختلال ژنتیکی نادر است که مشخصهاش میکروسفالی، کُند شدن روند رشد و تکامل بدن و چانهٔ عقبرفتهاست. علائم ممکن است از بیماری به بیمار دیگر فرق کند، اما برخی دیگر از نشانهها عبارتند از: صدای زیر و نازک، انگشتان پردهدار دست و پا، ناهنجاریهای کام، مشکلاتی در زبان تکلمی و دوری گزیدن از شلوغی و جمعیت.[2] نحوهٔ بیماریزایی سندرم دوبوویتز هنوز مشخص نشدهاست و هیچ روش آزمایشگاهی قطعی برای تشخیص آن وجود ندارد.[3] روش اصلی تشخیص این بیماری، فنوتیپ خاص چهره است. از زمان نخستین توصیف این بیماری در سال ۱۹۶۵ میلادی توسط پزشک انگلیسی ویکتور دوبوویتز، بیش از ۱۴۰ مورد از این بیماری در سرتاسر جهان شناسایی شدهاست. با آنکه بیشترین نمونهها از ایالات متحده آمریکا، آلمان و روسیه بودهاست، اما این بیماری در هر دو جنس مؤنث و مذکر و تمامی نژادها بهطور یکسان دیده میشود.[2]
| سندرم دوبوویتز | |
|---|---|
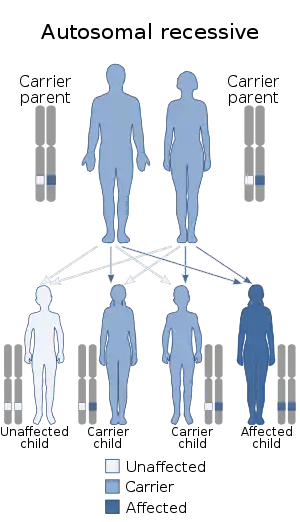 | |
| نحوهٔ وراثت این بیماری از نوعِ اتوزومال مغلوب است. | |
| تخصص | ژنشناسی پزشکی |
| علامتها | عقبافتادگی رشد داخل رحمی، قد کوتاه، میکروسفالی، عقبماندگی ذهنی خفیف و مشکلات رفتاری، اِگزما، مشخصههای غیرعادی و خاص چهرهای[1] |
| تشخیص پزشکی | تشخیص بر پایهٔ علائم و مشخصات ظاهری فرد است که توسط متخصص ژنتیک یا پزشک انجام میشود. |
| درمان | به سبب نادر بودنِ بیماری و فقدان هرگونه درمان قطعی، باید بهخاطر داشت که این افراد، رشد و تکامل کُندتری نسبت به بقیه دارند و اغلب نیازمندِ توجه خاص و مراقبتهای فردی هستند. مراقبتهای تکبهتک در مورد برخی از آنها بهتر است تا بتواند پاسخگوی نیازهای خاص آنان باشد. |
تظاهر بیماری
یکی از مشخصههای این بیماری میکروسفالی است که طی آن اندازهٔ دور سر کمتر از حد طبیعی است و علتش رشد کمتر بافت مغز است. میکروسفالی ممکن است به دلیل اختلالات ژنتیکی، عفونتها، پرتوتابی، مصرف برخی داروها و سوءمصرف الکل در دوران بارداری ایجاد شود. اختلال در رشد قشر مغز منجر به بروز بسیاری از علائم مربوط به میکروسفالی میگردد.[4]
پیشآگهی میکروسفالی بسیار متغیر است: برخی مبتلایان، کندذهنی خفیف و اندکی دارند و به شاخصههای رشدی موردِ انتظارِ سن خود میرسند. در برخی دیگر، کندذهنی شدید است و دچار عوارض عصبی-ماهیچهای نیز هستند.[4]
ژنتیک

با آنکه پاتولوژی سندرم دوبوویتز نامعلوم است، اما روش انتقال آن از نوع اتوزومال مغلوب است. والدین افراد مبتلا به این بیماری، گاهی نسبت فامیلی دارند و چندین مورد از آن در دوقلوهای تکتخمکی، خواهر-برادرها و خویشاوندی همنیایی (فرزند عمو، دایی، عمه یا خاله) دیده شدهاست.[5] گوناگونی فنوتیپی فراوانی میان بیماران، بهویژه در زمینهٔ هوش دیده میشود.[6] با آنکه شواهد فراوانی دالِ بر ژنتیکی بودن این سندرم موجود است، اما علائم آن بسیار شبیه به ناهنجاریهای جنینی ناشی از الکل است. پژوهشهای بیشتری لازم است تا مشخص است آیا الکل سبب ایجاد این بیماری در افرادی میشود که از لحاظ ژنتیکی زمینهٔ آنرا دارند.[7] مشخص شده که در این بیماری، شکستگی کروموزومی وجود دارد.[5]
هورمون رشد
سندرم دوبوویتز با کمبود هورمون رشد همراه است.[8] مبتلایان به این بیماری دچار عقبافتادگی رشد هستند. هورمون رشد توسط بخش جلویی غدهٔ هیپوفیز ترشح میشود و وظیفهٔ اصلی آن در دوران رشد و نمو، «افزایش قد» است. بقیهٔ وظایف بخش جلویی غدهٔ هیپوفیز شامل تنظیم عملکرد دستگاه ایمنی، حفظ کلسیم بدن، افزایش تودهٔ عضلانی بدن و تحریک نوگلوکززایی است. کمبود هورمون رشد معمولاً در اثر جهشهای ژنتیکی، ناهنجاریهای غدد هیپوتالاموس یا هیپوفیز در هنگام تکامل، یا آسیب به غدهٔ هیپوفیز رخ میدهد.[9] در سندرم دوبوویتز معمولاً جهشهای ژنی یا ناهنجاریهای رشد و تکاملِ ساختارهای مغزی، باعث کمبود هورمون رشد میشود. کمبود این هورمون در این بیماران، با میزان کمبودِ پادتنهای IgG همخوانی دارد که در این بیماران یافت میشود.[5]
ارتباط با سندرم اسمیت- لملی- اوپیتز
پژوهشگران مشغول تحقیق و بررسی شباهتهایی هستند که میان سندرم دوبوویتز و سندرم اسمیت- لملی- اوپیتز (SLOS) وجود دارد. این دو بیماری شباهتهای فراوانی دارند و فرضیهای دربارهٔ ارتباط میان آن دو مطرح شدهاست. دو مشخصهٔ «سندرم اسمیت- لملی- اوپیتز»، غلظت کمِ کلسترول خون و غلظت زیاد ۷-دهیدروکلسترول است. کلسترول برای برخی کارها و فرایندهای مهم بدن ضروری است؛ از جمله حفظ ساختار غشایی سلولها، رویانزایی و ساخت هورمونهای استروییدی و جنسی. اختلال در تولید یا حملونقل کلسترول شاید علتِ بهوجود آمدنِ بسیاری از علائمِ هر دو سندرمِ یادشده باشد. گرچه تعداد اندکی از مبتلایان به سندرم دوبوویتز دچار سطح غیرطبیعی کلسترول هستند، اما پژوهشگران مایلند بدانند که آیا این سندرم هم همچون «سندرم اسمیت- لملی- اوپیتز» به سبب نقصی در مسیرهای ساخت و تولید کلسترول ایجاد میشود یا خیر.[10]
آسیبشناسی سندرم دوبوویتز به سبب نادر بودن آن و گسترهٔ وسیعی از علائمش هنوز نامشخص است. تاکنون چندین پژوهش برای یافتن علتِ بیماری و نحوهٔ بروزش انجام شدهاست. طی یکی از این پژوهشها، علائم خاص دهانی در یک بیمارِ مبتلا بررسی شد.[11] در پژوهشی دیگر، ناهنجاریهای مغزی مورد مطالعه قرار گرفت، از جمله اختلال در تکامل جسم پینهای، هیپوفیز قدامیِ کمرشد و ساقهٔ مغزی که دارای هیپوفیز خلفی نابجا است.[12]
تشخیص
هیچگونه روش تشخیصی قطعی برای این بیماری وجود ندارد. تشخیص بیماری بر پایهٔ مشخصات و علائم ظاهری فرد انجام میشود.
درمان
در حال حاضر، هیچگونه درمان اختصاصی برای این بیماری موجود نیست. مدیریت درمان، از طریق درمانهای حمایتی است. علیالخصوص، هیچگونه روش درمانی خاصی برای اصلاح میکروسفالی وجود ندارد؛ اما چندین درمان علامتی هست که میتوان با آنها، جلوی عواقب میکروسفالی را گرفت. برخی از این درمانهای حمایتی عبارتند از: گفتاردرمانی، کاردرمانی و داروهای کنترلکنندهٔ صرع و بیشفعالی.[13]
همهگیرشناسی
ما بین سالهای ۱۹۶۵ تا ۲۰۱۸ میلادی، در حدود ۲۰۰ نمونه از این بیماری گزارش شدهاست.[14]
تاریخچه
این بیماری نخستین بار در سال ۱۹۶۵ میلادی، توسط پزشک انگلیسی ویکتور دوبوویتز گزارش شد.
منابع
- "Dubowitz syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2017-07-11.
- "Dubowitz syndrome". Encyclopedia of Genetic Disorders. Archived from the original on 2007-04-18.
- "Dubowitz Syndrome Support Network".
- Microcephaly Information Page در مؤسسه ملی اختلالات عصبی و سکته مغزی
- Online 'Mendelian Inheritance in Man' (OMIM) Rasmussen, Sonja A. Dubowitz Syndrome -223370
- Ilyina HG, Lurie IW (1990). "Dubowitz syndrome: possible evidence for a clinical subtype". Am. J. Med. Genet. 35 (4): 561–5. doi:10.1002/ajmg.1320350423. PMID 2185633.
- Mathieu M, Berquin P, Epelbaum S, Lenaerts C, Piussan C (December 1991). "[Dubowitz syndrome. A diagnosis not to be missed]". Arch. Fr. Pediatr. (به French). 48 (10): 715–8. PMID 1793348.
- Hirano T, Izumi I, Tamura K (1996). "Growth hormone deficiency in Dubowitz syndrome". Acta Paediatr Jpn. 38 (3): 267–9. doi:10.1111/j.1442-200x.1996.tb03484.x. PMID 8741320.
- Rieser, Patricia (1979). "Growth Hormone Deficiency". Archived from the original on 2007-04-24.
- Ahmad A, Amalfitano A, Chen YT, Kishnani PS, Miller C, Kelley R (1999). "Dubowitz syndrome: a defect in the cholesterol biosynthetic pathway?". Am. J. Med. Genet. 86 (5): 503–4. doi:10.1002/(SICI)1096-8628(19991029)86:5<503::AID-AJMG21>3.0.CO;2-Y. PMID 10508998.
- Chan KM, King NM (2005). "Dubowitz syndrome: report of a case with emphasis on the oral features". J Dent Child (Chic). 72 (3): 100–3. PMID 16568913.
- Oguz KK, Ozgen B, Erdem Z (2003). "Cranial midline abnormalities in Dubowitz syndrome: MR imaging findings". Eur Radiol. 13 (5): 1056–7. doi:10.1007/s00330-002-1580-2. PMID 12695828.
- "Microcephaly – Symptoms, Treatment and Prevention". The HealthCentralNetwork. Archived from the original on 2007-05-13.
- Innes AM, McInnes BL, Dyment DA (2018) Clinical and genetic heterogeneity in Dubowitz syndrome: Implications for diagnosis, management and further research. Am J Med Genet C Semin Med Genet
- مشارکتکنندگان ویکیپدیا. «Dubowitz syndrome». در دانشنامهٔ ویکیپدیای انگلیسی، بازبینیشده در ۳۰ اکتبر ۲۰۲۰.
پیوند به بیرون
| طبقهبندی | |
|---|---|
| منابع بیرونی |
|
| در ویکیانبار پروندههایی دربارهٔ سندرم دوبوویتز موجود است. |