مطالعه همخوانی سراسر ژنوم
مطالعهٔ همخوانی سراسر ژنوم (به انگلیسی: genome-wide association study) (کوتاهشده: GWAS جیدبلیواِیاس یا GWA study) در دانش ژنتیک یک بررسی سراسری ژنوم بر روی مجموعهای از تنوّعهای ژنتیکی فردی در افراد مختلف است. هدف این بررسی مقایسه، و نتیجهگیری، و در نهایت رسیدن به رابطههای مشترک، و گونهای همخوانی، همبستگی و وابستگی، میان وجود یک تنوع ژنتیکی، و بروز و ظهور یک ویژگی مشترک در بین دارندگان آن تفاوت ژنتیکیاست. این مطالعات معمولاً روی بررسی ارتباط بین همخوانی چندریختیهای تک-نوکلئوتیدی (SNP اسانپی) و ویژگیهایی مانند بیماریهای عمدهٔ انسانی متمرکز است اما دادههای بهدست آمده میتواند برای هر ارگانیسم زندهٔ دیگری نیز مورد استفاده قرار گیرد.
زمانی که بررسی (جیدبلیواِیاس) روی دادههای انسانی اعمال میشود، این بررسیها دیاناِیِ افراد شرکتکننده را که فنوتیپهای متفاوتی برای یک ویژگی یا بیماری دارند با هم مقایسه میکنند. شرکتکنندگان در یک مطالعهٔ (جیدبلیواِیاس) میتوانند کسانی با داشتن بیماری (موارد مشخص) و افراد همسانی بدون (کنترل سابقه)، یا میتوانند کسانی با فنوتیپهای متفاوتی برای یک (موردِ ویژه) باشند، مثلاً فشار خون. به این روش اول-فنوتیپ (phenotype-first) گفته میشود که در آن افراد ابتدا بر اساس ظاهر بالینی گروهبندی میشوند. شیوهٔ متقابل، روش اول-ژنوتیپ (genotype-first) مطرح است. بعد از این مرحله نمونهٔ ژنتیکی هر فرد؛ که همان دیانای است، استخراج میگردد، حال اگر تناوب یک آلل مربوط به یک تنوع خاص در گروه بیماران بهطور معنیداری متداولتر از گروه شاهد باشد، مطالعه آن تنوع را با بیماری همبسته (همخوان یا مرتبط) خواهد خواند؛ بنابراین چندریختیهای همبسته با بیماری برای نشانهگذاری نواحی مرتبط با بیماری استفاده میشوند. در این مطالعه به جای در نظر گرفتن نواحی محدودی در ژنوم که مستعد همبستگی با بیماری (یا ویژگی) هستند، کل ژنوم را در نظر میگیریم، بنابراین به این رویکرد، غیر-نامزد-محور (non-candidate-driven) میگوییم که در مقابل رویکرد نامزد-محور (candidate-driven) قرار میگیرد. مطالعات همخوانی سراسر ژنوم توانایی یافتن ژنهایی را که دلیل رخدادن بیماریها هستند ندارد، گرچه با این مطالعات میتوان تنوّعهای همبسته با بیماریها را تشخیص داد. (دقت کنید رابطهٔ علیّت هم ارز رابطه همبستگی نیست)[2][3][4]
نتایج اولین مطالعه موفق در سال ۲۰۰۵ منتشر شد. این مطالعه روی بیمارانی صورت گرفت که به تحلیلرفتن عضلانی مرتبط با سن (age-related macular degradation) دچار بودند، دو چندریختی (اسنیپ) یافت شد که به شکل معنیداری در تناوب آلل با گروه شاهد تفاوت داشت.[5]
از سال ۲۰۱۱ صدها یا هزاران نفر آزمایش شدهاند، بیش از ۱۲۰۰ مطالعه همخوانی سراسر ژنوم انسانی روی بیش از ۲۰۰ بیماری و صفت صورت گرفته و تقریباً ۴۰۰۰ همبستگی برای چندریختیها (اسنیپ) کشف شدهاند.[6] تعدادی از مطالعات با انتقاداتی مبنی بر عدم دقت در آزمایش همراه بودهاند، گرچه مطالعات جدید این مشکلات را مرتفع کردهاند. در هر حال روشهای مورد استفاده مخالفانی دارد.
پیش زمینه
هر دو ژنوم انسان، در میلیونها مورد با هم متفاوتند. این تفاوت به صورتهای مختلفی وجود دارد. تفاوتهای کوچک در تک نوکلئوتیدهای ژنوم که همان چندریختیها هستند، تفاوتهای بزرگتر مانند درجها، حذفها و تنوعهای کپی-تعداد هر کدام از این تغییرات میتوانند باعث دگرشکلیهایی در صفات یا فنوتیپ شوند که میتواند بیماری، یا هر صفت با نمود فیزیکی باشد.[8] در حدود سال ۲۰۰۰، پیش از معرفی مطالعه همخوانی سراسر ژنوم، روش اولیه تحقیق در این زمینه از طریق پیوند ژنتیکی در خانوادهها بود. این روش به عنوان یک روش بسیار کارا برای مطالعه اختلالات تک ژنی (اختلالاتی که تنها یک ژن در رخدادن آنها دخیل است) شناخته میشد.[9] در هر حال برای بیماریهای رایج و پیچیده، نتایج حاصل از مطالعات پیوند ژنتیکی به سختی قابل تعمیم بودند.[8][10] یک پیشنهاد جایگزین برای مطالعات پیوندی، مطالعه همبستگی ژنتیکی بود. این نوع مطالعه میپرسد آیا آلل یک تنوع ژنتیکی بیشتر از میزان مورد انتظار در افراد دارای فنوتیپ مورد علاقه یافت میشود یا نه. محاسبات اولیه روی توان آماری نشان دادند این رویکرد میتواند در تشخیص اثرهای ضعیف ژنتیکی بهتر عمل کند.[11]
علاوه بر چارچوب مفهومی، چندین عامل دیگر هم مطالعات همخوانی سراسر ژنوم را ممکن کردند. یکی از این عوامل ظهور بیوبانکهاست، که مخزن اطلاعات ژنتیکی انسان هستند که به میزان چشمگیری هزینه و دشواری جمعآوری تعداد کافی از نمونههای زیستی برای مطالعه را کاهش داد.[12] پروژههای زیستی بزرگ مانند پروژه بینالمللی هپمپ و پروژه ۱۰۰۰ ژنوم نیز با شناخت چندریختیهای جدید به کمک مطالعات همبستگی آمدند.[13][14]
روش
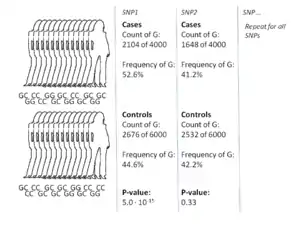
رایجترین رویکرد در مطالعه همخوانی سراسر ژنوم روش مورد-شاهد است که دو گروه بزرگ از افراد که یکی سالم (شاهد) و دیگری متأثر از یک بیماری (مورد) هستند را مقایسه میکند. ژنوتیپ همه افراد در هر گروه برای رایجترین اسنیپهای شناخته شدهاستخراج میشود. تعداد دقیق اسنیپها به تکنولوژی استخراج دادهها بازمیگردد، اما معمولاً این مقدار یک میلیون یا بیشتر است.[7]
برای هر کدام از این اسنیپها این آزمون انجام میشود که آیا تناوب آلل به شکل معنیداری بین دو گروه مورد و شاهد متفاوت است یا نه.[16] در چنین آزمونهایی واحد پایه برای بیان اندازه اثر، نسبت شانس (odds ratio) است. نسبت شانس نسبت دو شانس است که در زمینه مطالعه همبستگی سراسر ژنوم شانس بیماری برای افرادی با یک آلل خاص و شانس بیماری برای افراد بدون همان آللاند. زمانی که تناوب آل در گروه مورد (بیمار یا داری صفت خاص) بسیار بیشتر از گروه شاهد باشد، نسبت شانس بهطور معنیداری بیشتر از یک خواهد بود و این رویه برای تناوب کمتر برعکس است. علاوه بر این، معمولاً یک پی-مقدار (P-value) برای معنیدار بودن نسبت شانس است توسط یک آزمون خی-دو (chi-squared test) ساده بدست میآید. پیدا کردن نسبت شانسهایی که بهطور معنیداری با ۱ فاصله دارند هدف مطالعه سراسری ژنوم است زیرا این امر نمایانگر همبسته بودن اسنیپ (چندریختی) با بیماری خواهد بود.[16]
انواع گوناگونی از رویکرد مورد-شاهد موجود است. یک جایگزین رایج برای مطالعه همبستگی مورد شاهدی، تحلیل دادههای فنوتیپیک کمّی است، مثلاً قد یا غلطت زیستنشانها یا حتی بررسی میزان بیان ژنها. بهطور مشابه، آمارههای جایگزین که برای غالب (dominant) و مغلوب (recessive) طراحی شدهاند میتوانند استفاده شوند.[16] محاسبات مطالعه معمولاً با یک نرمافزار بیوانفورماتیکی مانند SNPTEST و PLINK انجام میشود که شامل انواع آمارهای قابل استفاده هستند[17][18]
مطالعات پیشین همبستگی سراسر ژنوم روی تک اسنیپها تمرکز میکردند. در حالی که آزمایشها نشان میدهند که برهمکنشهای پیچیدهای بین دو یا چند اسنیپ روی میدهد که ممکن است در رخدادن بیماریهای پیچیده دخیل باشد که به این پدیده اپیستاسیس (epistasis) میگویند. به علاوه، محققان سعی میکنند تا دادههای همبستگی سراسر ژنوم را با بقیه دادههای زیستی مثل شبکه برهمکنشهای پروتئین-پروتئین مجتمع کنند تا بتوان نتایج حاوی اطلاعات بیشتری را بدست آورد.[19][20]
یک گام کلیدی در اکثر مطالعات همخوانی سراسر ژنوم نسبتدادن ژنوتیپها به اسنیپها و نه به تراشه ژنوتیپ مورد استفاده در مطالعه است.[21] این روند تا حد زیادی تعداد اسنیپهایی که مورد آزمون میتوانند قرار گیرند را افزایش میدهد، توان مطالعه را بالا میبرد و فراتحلیل (meta analysis) را روی گروههای مختلف فراهم میکند. نسبتدادن ژنوتیپ با روشهای آماری که دادههای مطالعه را با یک منبع مرجع از هپلوتایپها ترکیب میکند، صورت میگیرد.
علاوه بر محاسبه همبستگی، رایج است که هر عامل که ممکن است بهطور بالقوه نتیجه را مخدوش کند گزارش شود. جنسیت و سن رایجترین عوامل مخدوشگر هستند. به علاوه، میدانیم که بسیاری از تمایزهای ژنتیکی با پیشینه تاریخی و جغرافیایی جوامع همبستهاند.[22] به دلیل این همبستگی، مطالعات باید پیشینه قومی و جغرافیایی شرکتکنندگان را گزارش کنند که به این فرایند تعیین قشر جامعه (population stratification) میگویند.
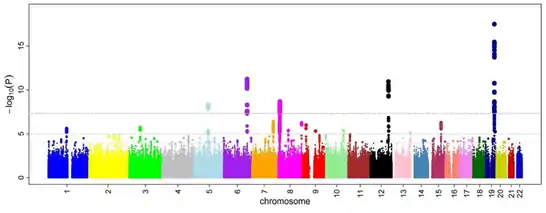
پس از آنکه نسبهای شانس و پی-مقدارها برای همه اسنیپها محاسبه شدند، یک رویکرد رایج رسم یک نمودار منهتن است. در زمینه مطالعات همبستگی ژنوم، این نمودار منفی لگاریتم پی-مقدار را به عنوان تابعی از جایگاه کروموزومی، نشان میدهد؛ بنابراین اسنیپهایی که با بالاترین سطوح همبستگی در نمودار مشخص میشوند. آستانه پی-مقدار برای سطح معنیدار بودن به خاطر مسائل چند-آزمونی تصحیح میشود. آستانه دقیق برای آزمایشهای مختلف متفاوت است،[23] اما معمولاً آستانه ۸-^۱۰ * ۵ برای سطح معنی داری در هر مقیاسی کار میکند.[7][16][24]
نتایج

تلاشهایی برای تهیه یک فهرست جامع از اسنیپهایی که در مطالعات همبستگی سراسر ژنوم شناخته شدهبودند انجام شدهاست.[26] از سال ۲۰۰۹، هزاران اسنیپ همبسته با بیماریها شناخته شدهاند.[6]
اولین مطالعه همخوانی سراسر ژنوم، در سال ۲۰۰۵ انجام شد که در آن ۹۶ بیمار با تحلیل عضلانی مربوط به سن (ARMD) با ۵۰ شاهد مقایسه میشدند.[27] در این مطالعه دو تا از اسنیپهایی که به شکل معنیداری تناوب آنها بین دو گروه متفاوت بود شناخته شدند. این اسنیپها روی فاکتور مکمل H قرار داشتند که یک دستآورد غیرمنتظره در مورد AMRD بود. یافتهها از این مطالعات اولیه به انجام پژوهشهای کاربردی بیشتر سرعت بخشید.[28] یکی دیگر از نقاط عطف تاریخ این مطالعات، مطالعه مورد-شاهدی کنسرسیوم ولکام تراست (WTCCC-Welcome Trust Case-Control Consortium) در سال ۲۰۰۷ بود که بزرگترین مطالعه همخوانی سراسر ژنوم است که تا به حال انجام شدهاست. این مطالعه شامل ۱۴٬۰۰۰ مورد (بیمار) مبتلا به هفت بیماری شایع (~۲٬۰۰۰ نفر برای هر کدام از عروق کرونر قلب، بیماریهای دیابت نوع ۱، با دیابت نوع ۲، با آرتریت روماتوئید، با بیماری کرون، با اختلال دو قطبی و فشار خون بالا) و ۳۰۰۰ مورد مشترک بود.[29] این مطالعه موفق به کشف بسیاری از ژنهای عامل این بیماریها شد.[29][30]
بعد از این مطالعات بسیار مهم اولیه، دو روند کلی وجود داشتهاست.[31] یکی روند بررسی نمونههای بزرگ و بزرگتر بودهاست. در پایان سال ۲۰۱۱، بزرگترین نمونهها در حدود ۲۰۰۰۰۰ نفر بود.[32] دلیل این رویکرد این است که بتوانیم نسبت به نتایج فرض شده مطمئنتر باشیم. روند دیگر استفاده از فنوتیپهای با تعریف دقیقتر مانند چربی خون، پروانسولین یا زیستنشانگرهای مشابه بود.[33][34] این فنوتیپها به فنوتیپهای حد واسط معروفند و تحلیل آنها میتواند برای پروژهشهای کاربردی روی زیستنشانگرها حائز اهمیت باشد.[35]
یک مسئله اساسی مورد بحث دربارهٔ مطالعات همخوانی سراسری ژنوم این بودهاست که اکثر تنوعهای چندریختی که توسط این مطالعات کشف شدهاند تنها با مقداری کمی از ریسک بیماری (یا صفت)، همبسته هستند و به میزان کمی در پیشبینیها مؤثرند. میانهٔ نسبت شانس به ازای هر اسنیپ مستعد ۱٫۳۳ است که تنها تعدادی از نسبتهای شانس بیشتر از ۰٫۳ هستند.[36][37] این مقادیر برای کشف تفاوتهای معنیدار کم به نظر میرسد زیرا میزن زیادی از تنوّع موروثی را توضیح نمیدهد. این تنوّع موروثی از تحقیقات موروثی روی دوقلوهای همسان به دست میآید.[38] به عنوان مثال مشخص شدهاست که ۸۰–۹۰٪ قد ارثی است، اما از این ۸۰–۹۰٪، مطالعات همبستگی تنها اقلیتی را گزارش میکند.[39]
موضوعات مربوط
- نگاشت همبستگی زیستی
- بیوانفورماتیک
- اپیدمیولوژی
- ژنومیک
- عدم تعادل پیوند
- ژنتیک جوامع
- ژنومیک مقایسهای
- فنوتیپ
- ژنوتیپ
- بیماریهای ژنتیکی
منابع
- Ikram MK; Sim X; Xueling S; et al. (October 2010). McCarthy, Mark I, ed. "Four novel Loci (19q13, 6q24, 12q24, and 5q14) influence the microcirculation in vivo". PLoS Genet. 6 (10): e1001184. doi:10.1371/journal.pgen.1001184. PMC 2965750. PMID 21060863.
- Manolio TA; Guttmacher, Alan E.; Manolio, Teri A. (July 2010). "Genomewide association studies and assessment of the risk of disease". N. Engl. J. Med. 363 (2): 166–76. doi:10.1056/NEJMra0905980. PMID 20647212.
- Pearson TA; Manolio TA (March 2008). "How to interpret a genome-wide association study". JAMA. 299 (11): 1335–44. doi:10.1001/jama.299.11.1335. PMID 18349094.
- "Genome-Wide Association Studies". National Human Genome Research Institute.
- Klein RJ; Zeiss C; Chew EY; Tsai JY; et al. (April 2005). "Complement Factor H Polymorphism in Age-Related Macular Degeneration". Science. 308 (5720): 385–9. doi:10.1126/science.1109557. PMC 1512523. PMID 15761122.
- Johnson AD; O'Donnell CJ (2009). "An Open Access Database of Genome-wide Association Results". BMC Med. Genet. 10: 6. doi:10.1186/1471-2350-10-6. PMC 2639349. PMID 19161620.
- Bush WS; Moore JH (2012). "Chapter 11: genome-wide association studies". PLoS Comput Biol. 8 (12): e1002822. doi:10.1371/journal.pcbi.1002822. PMC 3531285. PMID 23300413.
- Strachan T; Read A (2011). Human Molecular Genetics (4th ed.). Garland Science. pp. 467–495. ISBN 978-0-8153-4149-9.
- "Online Mendelian Inheritance in Man". Retrieved 2011-12-06.
- Altmüller J; Palmer LJ; Fischer G; Scherb H; et al. (November 2001). "Genomewide Scans of Complex Human Diseases: True Linkage Is Hard to Find". Am. J. Hum. Genet. 69 (5): 936–50. doi:10.1086/324069. PMC 1274370. PMID 11565063.
- Risch N; Merikangas K (September 1996). "The future of genetic studies of complex human diseases". Science. 273 (5281): 1516–7. doi:10.1126/science.273.5281.1516. PMID 8801636.
- Greely HT (2007). "The uneasy ethical and legal underpinnings of large-scale genomic biobanks". Annu Rev Genomics Hum Genet. 8: 343–64. doi:10.1146/annurev.genom.7.080505.115721. PMID 17550341.
- The International HapMap Project, Gibbs RA, Belmont JW, Hardenbol P, Willis TD, Yu F, Yang H, Ch'Ang L-Y, Huang W (December 2003). "The International HapMap Project". Nature. 426 (6968): 789–96. doi:10.1038/nature02168. PMID 14685227.
- Schena M; Shalon D; Davis RW; Brown PO (October 1995). "Quantitative monitoring of gene expression patterns with a complementary DNA microarray". Science. 270 (5235): 467–70. doi:10.1126/science.270.5235.467. PMID 7569999.
- Wellcome Trust Case Control Consortium (June 2007). "Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls". Nature. 447 (7145): 661–78. doi:10.1038/nature05911. PMC 2719288. PMID 17554300.
- Clarke GM; Anderson CA; Pettersson FH; Cardon LR; et al. (February 2011). "Basic statistical analysis in genetic case-control studies". Nat Protoc. 6 (2): 121–33. doi:10.1038/nprot.2010.182. PMC 3154648. PMID 21293453.
- "Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls". Nature. 447 (7145): 661–678. 2007. doi:10.1038/nature05911. PMC 2719288. PMID 17554300.
- Purcell S; Neale B; Todd-Brown K; Thomas L; et al. (September 2007). "PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses". Am. J. Hum. Genet. 81 (3): 559–75. doi:10.1086/519795. PMC 1950838. PMID 17701901.
- Ayati, Marzieh; Erten, Sinan; Chance, Mark R.; Koyutürk, Mehmet (2015-06-30). "MOBAS: identification of disease-associated protein subnetworks using modularity-based scoring". EURASIP Journal on Bioinformatics and Systems Biology. 2015 (1): 1–14. doi:10.1186/s13637-015-0025-6. ISSN 1687-4153.
- Ayati, Marzieh; Koyutürk, Mehmet (2015-01-01). "Assessing the Collective Disease Association of Multiple Genomic Loci". Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics. BCB '15. New York, NY, USA: ACM: 376–385. doi:10.1145/2808719.2808758. ISBN 978-1-4503-3853-0.
- Marchini J; Howie B (2010). "Genotype imputation for genome-wide association studies". Nature Reviews Genetics. 11 (7): 499–511. doi:10.1038/nrg2796. PMID 20517342.
- Novembre J; Johnson T; Bryc K; Kutalik Z; et al. (November 2008). "Genes mirror geography within Europe". Nature. 456 (7218): 98–101. doi:10.1038/nature07331. PMC 2735096. PMID 18758442.
- Wittkowski KM; Sonakya V; Bigio B; Tonn MK; et al. (January 2014). "A novel computational biostatistics approach implies impaired dephosphorylation of growth factor receptors as associated with severity of autism". Transl Psychiatry. 4 (1): e354. doi:10.1038/tp.2013.124. PMC 3905234. PMID 24473445.
- Barsh GS; Copenhaver GP; Gibson G; Williams SM (5 July 2012). "Guidelines for Genome-Wide Association Studies". PLoS Genetics. 8 (7): e1002812. doi:10.1371/journal.pgen.1002812. PMC 3390399. PMID 22792080.
- Sanna S, Li B; Mulas A; Sidore C; Kang HM; et al. (July 2011). "Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability". PLoS Genet. 7 (7): e1002198. doi:10.1371/journal.pgen.1002198. PMC 3145627. PMID 21829380.
- Hindorff LA; Sethupathy P; Junkins HA; Ramos EM; et al. (June 2009). "Potential etiologic and functional implications of genome-wide association loci for human diseases and traits". Proc. Natl. Acad. Sci. U.S.A. 106 (23): 9362–7. doi:10.1073/pnas.0903103106. PMC 2687147. PMID 19474294.
- Haines JL; Hauser MA; Schmidt S; Scott WK; et al. (2005). "Complement Factor H Variant Increases the Risk of Age-Related Macular Degeneration". Science. 308 (5720): 419–421. doi:10.1126/science.1110359. PMID 15761120.
- Fridkis-Hareli M; Storek M; Mazsaroff I; Risitano AM; et al. (October 2011). "Design and development of TT30, a novel C3d-targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway–mediated diseases". Blood. 118 (17): 4705–13. doi:10.1182/blood-2011-06-359646. PMC 3208285. PMID 21860027.
- Wellcome Trust Case Control Consortium, Burton PR; Clayton DG; Cardon LR; et al. (June 2007). "Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls". Nature. 447 (7145): 661–78. doi:10.1038/nature05911. PMC 2719288. PMID 17554300.
- "Largest ever study of genetics of common diseases published today" (Press release). Wellcome Trust Case Control Consortium. 2007-06-06. Retrieved 2008-06-19.
- Ioannidis JP; Thomas G; Daly MJ (2009). "Validating, augmenting and refining genome-wide association signals". Nat Rev Genet. 10 (5): 318–29. doi:10.1038/nrg2544. PMID 19373277.
- Ehret GB; Munroe PB; Rice KM; Bochud M; et al. (October 2011). "Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk". Nature. 478 (7367): 103–9. doi:10.1038/nature10405. PMC 3340926. PMID 21909115.
- Kathiresan S; Willer CJ; Peloso GM; Demissie S; et al. (January 2009). "Common variants at 30 loci contribute to polygenic dyslipidemia". Nat. Genet. 41 (1): 56–65. doi:10.1038/ng.291. PMC 2881676. PMID 19060906.
- Strawbridge RJ; Dupuis J; Prokopenko I; Barker A; et al. (October 2011). "Genome-Wide Association Identifies Nine Common Variants Associated With Fasting Proinsulin Levels and Provides New Insights Into the Pathophysiology of Type 2 Diabetes". Diabetes. 60 (10): 2624–34. doi:10.2337/db11-0415. PMC 3178302. PMID 21873549.
- Danesh J; Pepys MB (November 2009). "C-reactive protein and coronary disease: is there a causal link?". Circulation. 120 (21): 2036–9. doi:10.1161/CIRCULATIONAHA.109.907212. PMID 19901186.
- Maher B (November 2008). "Personal genomes: The case of the missing heritability". Nature. 456 (7218): 18–21. doi:10.1038/456018a. PMID 18987709.