سندرم جانسون بلیزارد
نشانگان جانسون بلیزارد (به انگلیسی :Johanson–Blizzard syndrome مخفف آن :JBS) یک بیماری نادر است که نحوه توارث آن به صورت اتوزوم مغلوب است و میتواند چندین ارگان در بدن را درگیر کند. در این بیماری لوزالمعده، جمجمه و گوش تکامل غیرطبیعی دارند و عقب ماندگی ذهنی، اختلال شنوایی و اختلال رشد با آن همراهی دارد.[1] بعضی این بیماری را نوعی دیسپلازی اکتودرمی در نظر میگیرند.[2]
| سندرم جانسون بلیزارد | |
|---|---|
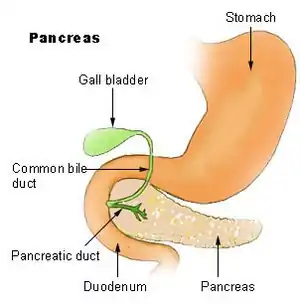 | |
| لوزالمعده و موقعیت آن در دستگاه گوارش. | |
| طبقهبندی و منابع بیرونی | |
| تخصص | ژنشناسی پزشکی |
| آیسیدی-۱۰ | Q45.0 |
| آیسیدی-9-CM | 751.7 |
| اُمیم | 243800 |
| دادگان بیماریها | 31914 |
اختلالات این بیماری بهطور به خصوصی اختلال شدید تکاملی و اختلال قسمت برونریز لوزالمعده را شامل میشود بهطوریکه گاهی اوقات این بیماری را یک بیماری لوزالمعدهای ارثی در نظر میگیرند.[3]
ویژگیها
غدد برونریز
مهمترین اختلال نشانگان جانسون بلیزارد نارسایی قسمت برونریز لوزالمعده میباشد.[1][4][5][6][7]
مهمترین نگرانی پس از تشخیص جانسون بلیزارد سوء هاضمه و هایپوگلایسمی است، کاهش ترشح لیپاز، تریپسین و تریپسینوژن باعث سوء جذب چربیها میشود. همچنین عدم ترشح گلوکاگون و افزایش فعالیت انسولین زمینه را برای هایپوگلایسمی فراهم میآورد.[1][3][8]
از جمله اختلالات در تکامل لوزالمعده میتواند مرگ برنامهریزی شده (آپوپتوز) ناقص، آسیب التهابی دورهٔ جنینی و پس از جنینی، نکروز و فیبروز آسینیهای پانکراس (قسمتی از لوزالمعده که ترشح و تولید شیرهٔ پانکراس در آن اتفاق میافتد) و جابجایی مادرزادی آسینیها با بافت چربی را میتوان نام برد.[1][3][8][9][10] جایگزینی کل پانکراس با بافت چربی تا به حال گزارش شده که میتواند کشنده باشد.
غده درونریز
نشانگان جانسون بلیزارد همچنین میتواند باعث نارسایی درون ریز (اندوکرین) پانکراس شود. نارسایی درون ریز نسبت به نارسایی برونریز شیوع کمتری دارد.[1]
جزایر لانگرهانس در پانکراس مجاریهایی هستند که وظیفهٔ تولید هورمونها را دارند و هورمونهایی نظیر انسولین، سوماتواستاتین و گلوکاگون را میسازد و در واقع بخش درونریز پانکراس را تشکیل میدهند. نارسایی درون ریز در سندرم جانسون بلیزارد میتواند به خاطر ساخته شدن چربی در جزایر لانگرهانس یا اختلال عصبدهی آن باشد.[1][5][8][11][12]
اختلال در بخش درونریز پانکراس میتواند ایجاد دیابت شیرین کند. هر دونوع دیابت وابسته به انسولین و دیابت کاهش تولید انسولین میتواند در سندرم جانسون بلیزارد دیده شود.[5][11]
تولید و ترشح الکترولیتها نظیر بیکربنات در پانکراس طبیعی است و سطوح آن نرمال باقی میماند.[1]
در این بیماری علاوه بر پانکراس، در بقیه غدد درونریز هم میتوانند اختلال داشته باشند و اختلالاتی مثل کمکاری تیروئید (هایپوتیروئیدیسم)،[2] کمبود هورمون رشد،[1][8] کمکاری هیپوفیز[1] ایجاد شود.
قسمت قدامی هیپوفیز میتواند دچار هامارتوم گلیال (به انگلیسی: glial hamartoma- تومور سلولهای گلیال) شود.[13] به دلیل سوء جذب چربیها، قسمت قدامی هیپوفیز میتواند نارسا شود که نتیجهٔ آن کاهش تولید هورمون رشد میباشد. این کاهش هورمون رشد میتواند باعث اختلال در رشد شود که نتیجهٔ آن کوتولگی (به انگلیسی: Dwarfism) میباشد.[1][4][14]
بینی
هایپوپلازی (عدم تکامل) پرههای بینی اولین بدشکلی اولیهٔ سندرم جانسون بلیزارد میباشد.[1][2][7] از جمله بدشکلیهای دیگر رایج در صورت افتادگی عضله پره بینی (alae nasi muscle)و عدم وجود غضروف بینی میباشد. این بدشکلیها در کل باعث میشوند سوراخهای بینی شکلی عجیب پیدا کند.[7][15]
عصبی
عقب ماندگی ذهنی از شدید تا ملایم در بیشتر این افراد وجود دارد. علت آن وجود یک مادهٔ زیانآور به نام جهشزا (موتاژن) است که در تکامل دستگاه عصبی مرکزی اختلال ایجاد میکند.[1][6][16] با این حال گزارش شده بسیاری از افراد دارای سندرم نشانگان بلیزارد دارای هوش طبیعی و تکامل اجتماعی مناسب سنشان داشتهاند.[12][16]
شنوایی
مطالعهای که روی گوش داخلی این بیماران انجام شد، فقدان شنوایی به دلیل گیرندههای عصبی (sensorineural hearing loss) در بسیاری از این بیماران را نشان داد. در این بیماران بافت کیستی در حلزون و دهلیز هر دو گوش تشکیل میشود که با اتساع این ساختارها باعث بدشکلی گوش میشود.[7][9][17] همچنین در این بیماران، بدشکلیهای مادرزادی استخوان گیجگاهی، اثرات سوءی بر میزان شنوایی این افراد دارد.[17][18]
جمجمه و صورت
در این بیماری آنومالیهای دیگری نیز ممکن است در نواحی مختلف جمجمه نظیر سر، صورت، فک و دندانها یافت شود که شامل: نقص اکتودرمی در خط وسط جمجمه با موهای کم پشت،[2][9] آپلازیا کوتیس (به انگلیسی: aplasia cutis- پوست خیلی نازک سر)،[19] فونتانل بزرگ (نقطهای نرم روی سر نوزادان)،[14] میکروسفالی،[19] برجستگی جلوی سر،[14] عدم وجود ابرو و مژه،[14] چشمهای شبه مغولی،[17] فیسچول مجرای بینی-اشکی (به انگلیسی:nasolacrimal duct fistula- که یک راهی بین مجرای اشکی و پوست صورت ایجاد میشود)،[9] گوشهای تخت،[14] کوچک بودن ماگزیلا(فک بالا) و مندیبل(فک پایین) که ماگزیلا بیشتر تحت تأثیر قرار میگیرد،[14][20][21] وجود شکاف مادرزادی در استخوان اوربیت (حدقه چشم)،[20] وجود اختلال در دندانهای شیری و عدم وجود دندانهای دائمی.[9][14]
دیگر ارگانها
علاوه بر آنومالیهای مادرزادی بالا، اختلال در دیگر ارگانها نیز وجود دارد که کمتر شایع است شامل: مقعد سوراخ نشده،[22] ریفلاکس حالبی مثانهای (وزیکواورترال، برگشت ادرار از مثانه به حالب و کلیهها)،[14] دوشاخه بودن مثانه و رحم نوزادان دختر،[7] کلستاز نوزادی کبد همراه با سیروز و هایپرتنشن (افزایش فشار) ورید پورت کبدی،[22] کاردیومیوپاتی متسع،[23] دکستروکاردی (سمت راست بودن مادرزادی قلب)،[1] نقص دیواره بطنی و دهلیزی (ASD,VSD)،[1] کم بودن وزن تولد (LBW)،[24] عدم پیشرفت،[24] هایپوتونی (کم بودن تون عضلانی)،[19] شکاف استخوان خاجی،[24] آب مروارید مادرزادی،[24] دانههای cafe-au-lait[2]
ژنتیک
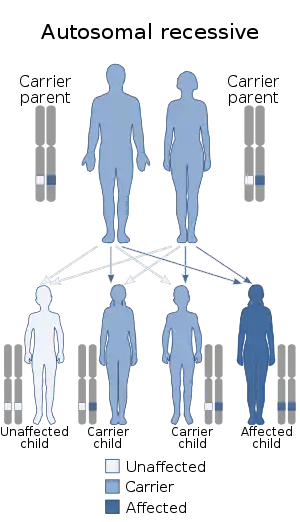
جانسون بلیزارد با یک الگوی اتوزوم مغلوب به ارث میرسد.[1] یعنی ژنی که مسئول ایجاد این سندرم است روی کروموزومهای اتوزوم قرار گرفته و برای اینکه نوزدای مبتلا باشد نیاز است دو کپی از آن ژن ناقص را در کروموزومهایش داشته باشد. والدین شخص بیمار یک کپی از این ژنهای معیوب را دارند ولی خودشان علائم بیماری را بروز نمیدهند.
مکانیسم بیماریزایی (پاتوفیزیولوژی)
علت نشانگان جانسون بلیزارد جهش در ژن UBR1 میباشد که مسئول کد کردن یکی از آنزیمهای متعدد یوبیکیتین لیگاز (ubiquitin ligase) است.[1][6]
پروتئین یوبیکیتین نامش از یوبیکوئیتوس (ubiquitous) به معنای همه جا در دسترس گرفته شدهاست چون در تمامی یوکاریوتها بیان میشود. یوبیکیتین یک پروتئین تنظیمی است که دیگر پروتئینها را با علامتدار کردن برای تخریب به وسیلهٔ پروتئوزوم تنظیم میکند.[25] وقتی یوبیکیتین لیگاز، مولکول یوبیکیتین را به لیزینِ زنجیره جانبی پروتئینهای هدف (که نقش سوبسترا بازی میکنند) متصل میکند پروسهٔ تنظیمی شروع میشود. این آنزیم بارها همین کار را تکرار میکند که به این فرایند پلی یوبیکیتینیشن (polyubiquitination) میگویند. این آنزیم پروئینهای آسیب دیده غیرطبیعی را با این فرایند تخریب میکند. فرایند پلی یوبیکیتینیشنِ پروتئینهای هدف به پروتئوزوم علامت میدهد تا آنها را تخریب کند که اینکار را با پروتئولیز انجام میدهد.[25] سیستم یوبیکیتین-پروتئوزوم نقش مهمی در دخریب غیر لیزوزومی پروتئینهای داخل سلولی بازی میکند. همچنین یوبیکیتین میتواند در اصلاح و ویرایش پروتئینها بعد از ترجمه نیز نقش داشته باشد.[25][26][27] تخریب و اصلاح پروتئینها در درون سلول فقط بخش کوچکی از سیستم تنظیمی داخل سلولی است که برای فرایندهایی نظیر تقسیم سلولی، نشاندار شدن سلولی، عملکرد گیرندههای سطح سلول، آپوپتوز، تعمیر دی ان ای، پاسخ التهابی و کیفیت کنترل مرتبط با چرخهٔ سلولی و هموستاز نیاز است[26][27]
تخریب پروتئینها با یوبیکیتین طبق قانون N آخر انجام میشود[28][29] در یوکاریوتها مثل انسان، مسیر Nآخر بخشی از سیستم یوبیکیتین را تشکیل میدهد.[28]
قانون N آخر میگوید اسیدآمینهای که در پایان پروتئینها وجود دارد یک گروه آمیدی (N) دارد که پروتئینها با این گروه آمیدی شناسایی میشوند. سیستم یوبیکیتین پروتئینهای ناکارآمد را با آن N آخر شناسایی کرده و تخریبشان میکند.[28][29][30]
در سندرم جانسون بلیزارد با جهش در ژن UBR1، یوبیکیتین لیگاز ساخته نمیشود.[1][6] در سلولهای آسینیهای پانکراس، UBR1 بیش از همه جای دیگر بدن بیان میشود.[1] کمبود فعالیت یوبیکیتین لیگاز باعث نقص در سیستم یوبیکیتین-پروتئوزوم میشود و خاصیت مرگ برنامهریزی شده (آپوپتوز) سلولهای آسیب دیده از بین میرود و اختلالاتی نظیر آسیب التهابی، جایگزینی بافت چربی، تکثیر بافت همبند در جزایر و آسینیهای پانکراس ایجاد میشود.[1][3][6] همچنین نقاط دیگر بدن نیز با همین مکانیسم تحت تأثیر قرار میگیرند مثل نواحی جمجمه و صورت، اسکلتی عضلانی، سیستم عصبی، دندانها و اعضا.[1][6][22]
موتاسیونهای بدمعنی (missense)، بیمعنی (nonsense) و خاموش(splice) UBR1 در هر دو والدین افراد سندرم جانسون بلیزارد یافت شده که هموزیگوس بودن فنوتیپ سندرم جانسون بلیزارد را تأیید میکند. تفاوتهای فنوتیپی در بیماران سندرم جانسون بلیزارد به دلیل وجود باقیماندههای یوبیکیتین لیگاز است که میتواند به دلیل جهشهای هیپومورفیک باشد که در والدین آنها دیده میشود.[1][3][6][22][23] ژن UBR1 در انسانها روی کروموزوم ۱۵ قرار دارد[6] .
درمان
با اینکه هیچ درمانی برای معالجه سندرم جانسون بلیزارد وجود ندارد ولی برای برطرف کردن بعضی از علامتهای آن درمان وجود دارد. براساس شدت سندرم جانسون بلیزارد، انتخاب نوع درمان برای برطرف کردن علائم در افراد مختلف متفاوت است.
نقص پانکراس و سوءجذب میتواند با تجویز آنزیمهای پانکراس مثل پانکرلیپاز (pancrelipase) و دیگر روشها برطرف شود.[1]
بدشکلیهای صورت و اسکلت میتواند با تکنیکهای جراحی مثل پیوند استخوان و استئوتومی ترمیم شود.[20] نقص شنوایی میتواند با سمعک و وسایل مشابه بهبود یابد.[12][17]
آموزش مخصوص، روشهای یادگیری دونفره، کاردرمانی میتواند وضعیت عقب ماندگی ذهنی آنها را بهتر کند تا هم برای پدر و مادر و هم برای جامعه مفید باشند.[31]
نامگذاری
دو متخصص اطفال یعنی آن جانسون (Ann J. Johanson) و رابرت بلیزارد (Robert M. Blizzard) این بیماری را برای اولین بار در سال ۱۹۷۱ توصیف کردند. برای همین به افتخار این دونفر این بیماری جانسون بلیزارد نام گرفت.[15][32]
منابع
- Alkhouri N, Kaplan B, Kay M, Shealy A, Crowe C, Bauhuber S, Zenker M (Nov 2008). "Johanson-Blizzard syndrome with mild phenotypic features confirmed by UBR1 gene testing". World Journal of Gastroenterology: WJG. 14 (44): 6863–6866. doi:10.3748/wjg.14.6863. PMC 2773884. PMID 19058315. Archived from the original (Free full text) on 18 February 2012. Retrieved 1 April 2015.
- Kulkarni ML, Shetty SK, Kallambella KS, Kulkarni PM (Dec 2004). "Johanson--blizzard syndrome". Indian Journal of Pediatrics. 71 (12): 1127–1129. doi:10.1007/BF02829829. PMID 15630323.
- Zenker M, Mayerle J, Reis A, Lerch MM (Jun 2006). "Genetic basis and pancreatic biology of Johanson-Blizzard syndrome". Endocrinology and Metabolism Clinics of North America. 35 (2): 243–253, vii–viii. doi:10.1016/j.ecl.2006.02.013. PMID 16632090.
- Sandhu BK, Brueton MJ (November 1989). "Concurrent pancreatic and growth hormone insufficiency in Johanson-Blizzard syndrome". J. Pediatr. Gastroenterol. Nutr. 9 (4): 535–8. doi:10.1097/00005176-198911000-00026. PMID 2621533.
- Steinbach WJ, Hintz RL (Nov 2000). "Diabetes mellitus and profound insulin resistance in Johanson-Blizzard syndrome". Journal of Pediatric Endocrinology & Metabolism: JPEM. 13 (9): 1633–1636. doi:10.1515/jpem.2000.13.9.1633. ISSN 0334-018X. PMID 11154160.
- Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, Beier M, Hülskamp G, Guzman C, Rehder H, Beemer FA, Hamel B, Vanlieferinghen P, Gershoni-Baruch R, Vieira MW, Dumic M, Auslender R, Gil-Da-Silva-Lopes VL, Steinlicht S, Rauh M, Shalev SA, Thiel C, Ekici AB, Winterpacht A, Kwon YT, Varshavsky A, Reis A (Dec 2005). "Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome)". Nature Genetics. 37 (12): 1345–1350. doi:10.1038/ng1681. PMID 16311597.
- Rosanowski F, Hoppe U, Hies T, Eysholdt U (Oct 1998). "Johanson-Blizzard syndrome. A complex dysplasia syndrome with aplasia of the nasal alae and inner ear deafness". HNO. 46 (10): 876–878. doi:10.1007/s001060050328. PMID 9846268.
- Takahashi T, Fujishima M, Tsuchida S, Enoki M, Takada G (Aug 2004). "Johanson-blizzard syndrome: loss of glucagon secretion response to insulin-induced hypoglycemia". Journal of Pediatric Endocrinology & Metabolism: JPEM. 17 (8): 1141–1144. doi:10.1515/jpem.2004.17.8.1141. ISSN 0334-018X. PMID 15379429.
- Daentl DL, Frías JL, Gilbert EF, Opitz JM (1979). "The Johanson-Blizzard syndrome: case report and autopsy findings". American Journal of Medical Genetics. 3 (2): 129–135. doi:10.1002/ajmg.1320030203. PMID 474625.
- Jones NL, Hofley PM, Durie PR (Sep 1994). "Pathophysiology of the pancreatic defect in Johanson-Blizzard syndrome: a disorder of acinar development". The Journal of Pediatrics. 125 (3): 406–408. doi:10.1016/S0022-3476(05)83286-X. PMID 8071749.
- Nagashima K, Yagi H, Kuroume T (Feb 1993). "A case of Johanson-Blizzard syndrome complicated by diabetes mellitus". Clinical Genetics. 43 (2): 98–100. doi:10.1111/j.1399-0004.1993.tb04458.x. ISSN 0009-9163. PMID 8448911.
- Gould NS, Paton JB, Bennett AR (Jun 1989). "Johanson-Blizzard syndrome: clinical and pathological findings in 2 sibs". American Journal of Medical Genetics. 33 (2): 194–199. doi:10.1002/ajmg.1320330212. PMID 2669481.
- Hoffman WH, Lee JR, Kovacs K, Chen H, Yaghmai F (Jan 2007). "Johanson-Blizzard syndrome: autopsy findings with special emphasis on hypopituitarism and review of the literature". Pediatric and Developmental Pathology: the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 10 (1): 55–60. doi:10.2350/06-05-0085.1. PMID 17378628.
- Fichter CR, Johnson GA, Braddock SR, Tobias JD (January 2003). "Perioperative care of the child with the Johanson-Blizzard syndrome". Paediatric Anaesthesia. 13 (1): 72–5. doi:10.1046/j.1460-9592.2003.00957.x. PMID 12535044.
- Online 'Mendelian Inheritance in Man' (OMIM) 243800
- Moeschler JB, Polak MJ, Jenkins JJ, Amato RS (January 1987). "The Johanson-Blizzard syndrome: a second report of full autopsy findings". Am. J. Med. Genet. 26 (1): 133–8. doi:10.1002/ajmg.1320260120. PMID 3812553.
- Braun J, Lerner A, Gershoni-Baruch R (1991). "The temporal bone in the Johanson-Blizzard syndrome. A CT study". Pediatric Radiology. 21 (8): 580–3. doi:10.1007/BF02012603. PMID 1815181.
- Bamiou DE, Phelps P, Sirimanna T (March 2000). "Temporal bone computed tomography findings in bilateral sensorineural hearing loss". Arch. Dis. Child. 82 (3): 257–60. doi:10.1136/adc.82.3.257. PMC 1718255. PMID 10685935.
- Mardin MK, Ghandour M, Sakati NA, Nyhan WL (Nov 1978). "Johanson-Blizzard syndrome in a large inbred kindred with three involved members". Clin Genet. 14 (5): 247–250. PMID 709902.
- Kobayashi S, Ohmori K, Sekiguchi J (Sep 1995). "Johanson-Blizzard syndrome facial anomaly and its correction using a microsurgical bone graft and tripartite osteotomy". J Craniofac Surg. 6 (5): 382–385. doi:10.1097/00001665-199509000-00011. PMID 9020718.
- Motohashi N, Pruzansky S, Day D (1981). "Roentgencephalometric analysis of craniofacial growth in the Johanson-Blizzard syndrome". J Craniofac Genet Dev Biol. 1 (1): 57–72. PMID 7341643.
- Al-Dosari MS, Al-Muhsen S, Al-Jazaeri A, Mayerle J, Zenker M, Alkuraya FS (July 2008). "Johanson-Blizzard syndrome: report of a novel mutation and severe liver involvement". Am J Med Genet A. 146A (14): 1875–9. doi:10.1002/ajmg.a.32401. PMID 18553553.
- Elting M, Kariminejad A, de Sonnaville ML, Ottenkamp J, Bauhuber S, Bozorgmehr B, Zenker M, Cobben JM (December 2008). "Johanson-Blizzard syndrome caused by identical UBR1 mutations in two unrelated girls, one with a cardiomyopathy". Am J Med Genet A. 146A (23): 3058–61. doi:10.1002/ajmg.a.32566. PMID 19006206.
- Dumić M, Ille J, Bobonj G, Kordić R, Batinica S (May 1998). "Johanson-Blizzardov sindrom". Lijec Vjesn (به Croatian). 120 (5): 114–6. PMID 9748788. Unknown parameter
|trans_title=ignored (help) - Wang J, Maldonado MA (August 2006). "The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases". Cell Mol Immunol. 3 (4): 255–61. PMID 16978533.
- Ciechanover A (September 1994). "The ubiquitin-mediated proteolytic pathway: mechanisms of action and cellular physiology". Biol Chem Hoppe-Seyler. 375 (9): 565–81. doi:10.1515/bchm3.1994.375.8.565. PMID 7840898.
- Ciechanover A, Iwai K (April 2004). "The ubiquitin system: from basic mechanisms to the patient bed". IUBMB Life. 56 (4): 193–201. doi:10.1080/1521654042000223616. PMID 15230346.
- Varshavsky A (January 1997). "The N-end rule pathway of protein degradation". Genes Cells. 2 (1): 13–28. doi:10.1046/j.1365-2443.1997.1020301.x. PMID 9112437.
- Baker RT, Varshavsky A (February 1991). "Inhibition of the N-end rule pathway in living cells". Proc Natl Acad Sci U.S.A. 88 (4): 1090–4. doi:10.1073/pnas.88.4.1090. PMC 50962. PMID 1899923.
- Gonda DK, Bachmair A, Wünning I, Tobias JW, Lane WS, Varshavsky A (October 1989). "Universality and structure of the N-end rule". J Biol Chem. 264 (28): 16700–12. PMID 2506181.
- Prater JF, D'Addio K (March 2002). "Johanson-Blizzard syndrome--a case study, behavioral manifestations, and successful treatment strategies". Biol Psychiatry. 51 (6): 515–7. doi:10.1016/S0006-3223(01)01337-3. PMID 11922888.
- Johanson A, Blizzard R (December 1971). "A syndrome of congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absent permanent teeth, and malabsorption". J Pediatr. 79 (6): 982–7. doi:10.1016/S0022-3476(71)80194-4. PMID 5171616.