لکودیستروفی متاکروماتیک
لکودیستروفی متاکرومیک (MLD) که کمبود آریل سولفاتاز A هم خوانده میشود، نوعی بیماری ذخیرهای لیزوزومی است.
| لکودیستروفی متاکروماتیک | |
|---|---|
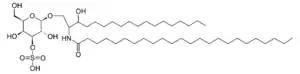 | |
| Sulfatide | |
| طبقهبندی و منابع بیرونی | |
| تخصص | غدد درون ریز و متابولیسم، عصبشناسی |
| آیسیدی-۱۰ | E75.2 |
| آیسیدی-9-CM | 330.0 |
| اُمیم | 250100 |
| دادگان بیماریها | 8080 |
| مدلاین پلاس | 001205 |
| ئیمدیسین | ped/۲۸۹۳ |
| سمپ | D007966 |
علائم بالینی
- نوع شیرخوارگی دیررس: قبل از ۳۰ ماهگی شروع شده و در عرض ۱ تا ۷ سال منجر به مرگ میشود. اولین تظاهر آن از دست دادن مهارتهای حرکتی اکتسابی بخصوص راه رفتن است. کاهش تونوس عضلانی، ژنو رکرواتوم شدید، کاهش یا غیاب رفلکسهای تاندونی، تأخیر در راه رفتن در برخی از بیماران، بروزعدم تعادل و ضعف بدنبال عفونت که ممکن است ناپدید شده و دوباره بعداً ظاهر شود و درد شدید پاها در بیمار رخ میدهد. در مرحله بعدی، عدم تعادل، دیزآرتری یا عدم تکلم، پسرفت عملکردهای مغزی، افزایش تونوس عضلانی و رفلکسهای تاندونی، نیستاگموس و آتروفی عصب بینایی و گاهی لکه قرمز آلبالویی در ماکولا ظاهر میشوند. سپس بیمار دچار فلج اسپاستیک همه اندامها، رژیدیتی دسربره یا رژیدیتی دکورتیکه، حرکات دیستونیک، تشنج در یک سوم موارد، اشکال در تغذیه و از دست دادن قدرت تکلم میشود. در مرحله آخر عدم پاسخ به والدین و لبخند زدن، از دست دادن ارتباط با محیط اطراف، کوری، عدم توانایی بلع و نیاز به تغذیه با لوله بینی-معدی بروز میکند و مرگ بیمار بدنبال ابتلا به ذات الریه رخ میدهد.
- نوع نوجوانی: بین ۴ تا ۱۶ سالگی با کاهش عملکرد در مدرسه، گاهی رفتار غیر معمول، حالت گیجی، مانس، سایکوز یا بیماری عاطفی، اختلال در راه رفتن، عدم تعادل، رژیدیتی عضلانی و اشکال در وضعیت بدن، عدم راه رفتن، بیاختیاری ادرار، تظاهرات احشایی غیرمعمول نظیر کوله سیستیت حاد، پانکراتیت خونریزی دهنده مزمن، توده شکمی یا خونریزی گوارشی بروز میکند.
- نوع بزرگسالی: پس از بلوغ حدود ۱۵ سالگی و گاهی در ۶۲ سالگی تظاهر میکند. در این نوع علائم روانی، دمانس به صورت از دست دادن حافظه یا کاهش توانایی ذهنی، اسکیزوفرنی، بیثباتی هیجانی، اضطراب یا بی تفاوتی، توهمات شنوایی، هذیان، سایکوز، افسردگی و الکلیسم مزمن مشاهده شدهاست. اشکالات حرکتی به صورت اختلال در راه رفتن و دیزآرتری، کاهش تونوس عضلانی و افزایش
رفلکسهای تاندونی، عدم تعادل و یافتههای مشابه پارکینسون، آسیب اعصاب محیطی در برخی از بیماران، دیستونی، ضعف اسپاستیک همه اندامها، آتروفی عصب بینایی و نیستاگموس، تشنج و وضعیت دکورتیکه در بیمار روی میدهند و سرانجام کوری، عدم تکلم و عدم پاسخ بروز میکند.
نقص آنزیم
نقص در آنریم آریل سولفاتاز A (سولفاتیداز)
توارث ژنتیکی
میزان بروز
نادر.
روش تشخیص
- مایع مغزی نخاعی: افزایش پروتئین.
- ادرار: افزایش سولفاتیدها.
- بررسی آنزیم: در فیبروبلاستها و گلبولهای سفید.
درمان
درمان حمایتی است. پیوند مغز استخوان در دوره قبل از بروز علائم یا اوایل آن ممکن است مفید باشد. پیوند در انواع نوجوانی و بزرگسالی به ترتیب نتایج خوب و تاحدی مفید داشتهاست، اما برای نوع شیرخوارگی دیررس توصیه نمیشود. آزمون تشخیص قبل از تولد: بررسی آنزیم در آمنیوسیتهای کشت شده و نمونه پرز کوریونی. نکته: در نوع کاذب نقص آنزیم، میزان آنزیم طبیعی است، اما فعالکننده ساپوسینB که کوفاکتوری است که فعالیت آنزیم را چند برابر افزایش میدهد، فعالیت کمی دارد.
جستارهای وابسته
منابع
- Poeppel P, Habetha M, Marcão A, Büssow H, Berna L, Gieselmann V (March 2005). "Missense mutations as a cause of metachromatic leukodystrophy, Degradation of arylsulfatase A in the endoplasmic reticulum". FEBS J. 272 (5): 1179–88. doi:10.1111/j.1742-4658.2005.04553.x. PMID 15720392
- بیماریهای متابولیک مادرزادی علائم بالینی، تشخیص و درمان، دکتر پریچهر توتونچی-امیر ساسان توتونچی